


|
|
DOI Prefix 10.20431 |
Information
Journal Policies
Orbital Rhabdomyosarcoma in an Adult, Case Report
Carlos A. Narvar Mendiola, Estephania Feria-Anzaldo, Medina Andrade Abraham Alejandro*, Grecia Yael Ortiz Ramirez, Ariel Ceriotto Garcia
Copyright : © 2017 . This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
1. Introduction
Rhabdomyosarcoma (RMS) is a highly malignant pluripotent mesenchymal tumor [1]. It is the most common soft-tissue sarcoma of the head and neck in childhood and constitutes about 4% of all pediatric malignancies, with 10% of all cases occurring in the orbit[1, 2]. Most of these tumors presents in the first decade of life[3], but RMS has been reported from birth to the eighth decade [1, 4, 5]. The incidence of malignant orbital tumors in general has been increasing in the last decades.6 Turner et al. in a population studyof incidence and survival in head and neck RMS reported an annual percentage increase of 1.16% and a statistically un- changed 5-year survival over the past 30years despite advances in treatment modalities; all this data against other studies with reports of improved survival [3]. Relative survival (RS) was more dependent on the extent of disease than primary site, and it was noted that most orbital RMS tumors (60.6%) presented with localized disease and, consequently, had a 5-year survival of about 84.3% which is more promising when compared with other sites in the head and neck[3].
2. Case Report
A 39-year-old female with progressive painful proptosis of 1 year of evolution come to external consult. She had temporal inferior displacement of her right eye and also presents exotropia and glance up limitation (Figure. 1); she refers vertical diplopia. Patient comments to have experienced weight loss the last two months and at physical exam a retroauricular lymphadenopathy was detected. Her best corrected visual acuity was 20/70 in the right eye and 20/40 in the left eye.
Left eye examination was normal; Exophthalmometry reading was 22 mm and 17 mm for right and left eye respectively.
She has history of an orbital biopsy 9 years prior to this event with histological diagnosis of a non-classified low-grade malign neoplasm.Orbital CT scan demonstrates an isodense and ill-circumscribed lesion of 5 x 4 cm, arising from the orbital roof without bone erosion (Figure. 2).


With the confirmation of an orbital tumor by imaging an anterior orbitotomy was performed through the medial upper eyelid crease and a violaceous well defined tumor was found and resected, with T2N0M0 classification.
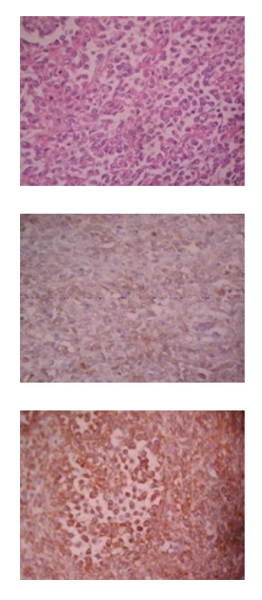
The histopathological study revealed a tumor composed of ill-defined aggregates of poorly differentiated oval tumor cells that show central loss of cellular cohesion, with scarce eosinophilic cytoplasm, round nucleus and formation of some irregular “alveolar” spaces. The neoplastic cells are aggregates of densely packed groups or masses of tumor cells resembling the areas of embryonal rhabdomyosarcoma, yet with a more uniform cellular picture with little or no fibrosis. The immunohistochemistry revealed immunoreactivity for vimentin, Myo-D1 and no immunoreactivity for S-100 protein and desmine. A solid variant of alveolar rhabdomyosarcoma was diagnosed. (Figure. 3)
The patient was referred to an oncological hospital with a III degree tumor classification by the IRSG. The patient underwent total orbital resection and was referred for radiotherapy and chemotherapy.
3. Discussion
RMS is a high-grade neoplasm of mesenchymal origin that arises from primitive skeletal muscle cells. It is a common soft-tissue malignancy in children and adolescents but infrequent in adults [1].
The orbit is the primary site in approximately 10% of children with RMS [2,4], and in adults orbital involvement is very rare [2].
The histopathologic types of RMS include embryonal, alveolar, pleomorphic and botryoide. Alveolar RMS is an aggressive subtype with a distinct histology, characterized by small round cells and with a poorer prognosis [1].
Alveolar RMS may present with a variety of histopathologic patterns. In the prevailing pattern, alveolar tumor cells are loosely adherent to a network of thin interstitial fibrovascular septa. The tumor cells are loosely attached to the connective tissue and in many areas become freely floating in the alveolar spaces [4,5].
Metastasis and recurrence of RMS in adults is associated with an unfavorable prognosis [1,4]. Moon et al. described a patient with ethmoidal sinus alveolar RMS of with infiltration to the orbit, and cervical lymph nodes involvement, who was treated with surgery and chemo-radiotherapy. The patient died after seven months secondary to metastasis to the spine [2]. Torres-Pena et al., described two adults with alveolar RMS, one patient had involvement of ethmoid sinus and right orbit; and the other patient presented tumor on the nasal cavity that infiltrates the orbit and the fronto-ethmoidal sinuses. Both patients died of systemic complications associated with the invasion.4 we report an atypical case of a RMS, with presentation in the orbit as primary site and without metastasis in the moment of diagnosis, which can improve the prognosis after chemotherapy and radiotherapy.
4. Conclusion
Although the established protocols for the treatment of RMS target the pediatric population, the diagnosis and treatment protocols in adults need to be extended by the rarity of this case. The prognosis of RMS greatly depends on prompt diagnosis and treatment.
References
- Wu T-H, Huang J-S, Wang H-M, Wang C-H, et. al. Long-term survivors of adult rhabdomyosarcoma of maxillary sinus following multimodal therapy: case reports and literature reviews. Chang Gung Med J. 2010;33(4):466– 71.
- Moon HS, Kwon SW, Lee JH. A case of alveolar rhabdomyosarcoma of the ethmoid sinus invading the orbit in an adult. Korean J Ophthalmol. 2006;20(1):70–5.
- Torres-Peña JL, Castrillo AIR, Mencía-Gutiérrez E, Gutiérrez-Díaz E, et.al. Nasal Cavity or Alveolar Paranasal Sinus Rhabdomyosarcoma with Orbital Extension in Adults: 2 Cases. PlastReconstrSurg Glob Open. Wolters Kluwer Health; 2015;3(6).
- Bagdonaite L, Jeeva I, Chang BYP, Kalantzis G, et.al. Multidisciplinary Management of Adult Orbital Rhabdomyosarcoma*. Orbit. Taylor & Francis. 2013;32(3):208–10.
- Sanz-Marco E, España E, Alamar A, Pérez- Rojas J, et.al. Orbital alveolar rhabdomyosarcoma masked by ethmoid sinusitis in a 25-year- old. Arch la Soc Española Oftalmol. English Ed. Elsevier; 2014; 89(5):1825.