


|
|
DOI Prefix 10.20431 |
Information
Journal Policies
Saint Christopher: Protection of Immunocompromised Travelers by Vaccination
Ger T. Rijkers*, Laura I.E. Yousif, Frans J. van Overveld
Copyright : © 2018 . This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Immunocompromised travelers are a growing population. Due to the advanced level of medicine, patients with chronic autoimmune diseases now for the first time have their diseases under control, are in better physical shape, and have greatly improved quality of life. As a result of this, these and other immunocompromised patients are now able to travel abroad should they want to. However, because of the immunocompromised condition, traveling comes with a risk for (severe) infectious diseases. Adequate and tailor made travel vaccination therefore is warranted. In this p aper an overview of the various types of vaccines and their mode of action is given. The current situation and prospects for the future of vaccination for immunocompromised travelers are being discussed.
1. Introduction
In 1969, the Catholic Church reviewed the calendar of Saints and discovered that for many of the Saints, including St. Christopher, there was absolutely no evidence of their existence. As a consequence, St. Christopher, among many others was dropped from the list [1]. However, this action did little to dampen the popularity of St. Christopher at all. The many believers of St. Christopher, when going on vacation, a business trip, or a real adventure, still invoke his blessings and protection as the Patron Saint of travelers. It is difficult indeed to end a tradition and belief that has been around for centuries (Figure 1).
Figure 1. Detail of the painting Saint Christopher by Jheronimus Bosch (approximately 1500), Museum Boijmans Van Beuningen, Rotterdam. The walking stick in the hand of Saint Christopher clearly has the configuration of an antibody molecule, symbolizing the protective effect of vaccination for travelers.

The function of the immune system is to defend the body against infections. It does this by recognizing, attacking and eliminating potentially infectious microorganisms. During an immune response, not only effector molecules and –cells are formed (antibodies and cytotoxic T cells, respectively) but also memory T- and memory B-lymphocytes. Upon repeated exposure to the same micro-organisms, the memory cells are reactivated and generate a so- called memory response, which is faster and stronger than a primary response. When the immune system would fail or is insufficient, there is an increased risk of severe infection, depending on the pathogenicity of the microorganism involved [3]. Vaccines therefore are being used with the purpose of strengthening the immune system by inducing protective antibodies and cytotoxic T cells, as well as specific immunological memory. The immunization schedules are designed with the aim to protect the whole population (including the most vulnerable) from being infected by major pathogens [4]. In addition, vaccination is important to limit the spread of a micro-organism in the population (herd immunity).
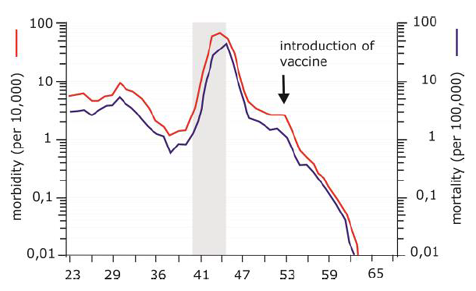
Vaccination is the best known and by far the most successful application of immunological principles in healthcare. The method of active immunization (see below) has reduced the incidence of target diseases by 95% or more (Figure 2), and in some cases such as smallpox,even complete eradication [5]. The word vaccine is derived from vaccinia, the cowpox virus (which was derived from the word la vache, French for cow). Edward Jenner was more than 200 years ago the first one to demonstrate that an infectious disease (in this case smallpox) can be prevented by vaccination [6]. A vaccine is defined as a product that induces an immune response in humans (or animals) to a specific disease without causing the clinical symptoms of the disease.
Figure 2. Vaccination against diphtheria has resulted in the virtual disappearance of the disease. Before World War II, diphtheria caused disease in 1-10 per 10,000 children in a European country as The Netherlands. Diphtheria is caused by Coryne-bacterium diphtheriae and its toxin affects the airways, heart and nervous system. In between 5 to 10% of the patients, the disease is fatal. The incidence of diphtheria peaked during World War II (indicated by the grey bar), with 60,400 cases in 1944. Introduction of mass vaccination in 1953 for all children between 0 and 5 years old lead to a sharp decline and virtual disappearance of diphtheria. Data from the National Institute of Public Health and the Environment (www.rivm.nl).
Consequently, the vaccinated individual becomes better protected against the infectious agent in question. It is important to realize that the first vaccines were developed in a period of time where knowledge about the function of the immune system or evidence based medicine was very limited.
Two forms of vaccination can be distinguished: passive vaccination and active vaccination. With passive vaccination, antibodies are administered. In active vaccination the individual is protected by inducing humoral and/or cellular immunity by administration of the vaccine [7]. The objective of active vaccination is to stimulate the immune system in such a way that, when coming into contact with a pathogenic micro-organism, a memory immune response is generated so quickly and efficiently, that infection is prevented.
2.The Des Ired Mode of Action of A Vaccine is Determined By The Pathogenes is of The Infection
The optimal mode of action of a vaccine is determined by the mechanism by which the micro-organism in question causes disease. The approach therefore will be different for micro- organisms that cause disease by the production of exotoxins (for example Clostridium tetani, Corynebacterium diphtheriae) or for micro- organisms that have to penetrate tissue (for example Neisseria meningitidis) or cells (measles virus) to cause infection or disease. The pathogenesis of a disease caused by a specific micro-organism also determines whether or not action should be taken to prevent colonization or to prevent infection. In the case of preventing colonization, the vaccine should induce a strong mucosal immune response that carriage of the micro-organism is prevented (see also below for polysaccharide vaccines).
In addition to knowledge of the pathogenesis of a microorganism causing disease, it is also necessary to know what constitutes a protective immune response [8]. This is because different types of microorganisms (e.g. bacteria, viruses, fungi) elicit completely different immune responses. As a rule of thumb, bacterial infections generally lead to a humoral immune response, while viral infections induce a cellular immune response. In many cases (such as influenza virus infections) both types of immune responses are necessary [9]. Furthermore, a distinction between the terms 'immunogenicity' and 'effectiveness' of a vaccine needs to be made. Immunogenicity is the ability of a vaccine to elicit an immune response in a target population [10]. Effectiveness is the ability of a vaccine to protect against a disease. Since the demonstration of the effectiveness of a vaccine requires large and lengthy studies, it would be helpful if the effectiveness of a vaccine could be based on the immune response elicited by the vaccination (this is termed "correlate or protection") [11].
3. Active Immunization Aims To Strengthen The Immune System
Active immunization is aimed to strengthening host defense and confer protection against infectious agents. That is why a vaccine ideally would closely mimic an in vivo immune response to the microorganism and in doing so stimulate a response of both the humoral as well as the cellular immune system.
Active immunization offers a number of benefits in comparison to passive immunization: it provides decades of protection, depending on the vaccine it can even be lifelong protection. Due to the induction of memory cells, protection persists even when the vaccine itself has already disappeared from the body [12]. Active immunization is relative inexpensive and thereby one of the most cost-effective measures in the healthcare. Passive immunization, which is the direct administration of specific antibodies, has the advantage that it offers immediate protection, which however is of limited duration. Passive immunization with in vitro generated human antibodies is considered for Ebola [13].
Most, not all, vaccines are administered during the beginning of life. In utero, exposure of the fetus to microorganisms is limited, and therefore the immune system develops in the absence of antigen-specific stimulation [14]. Directly after birth, infants are exposed to a sheer limitless number of (potential pathogenic) microorganisms from the environment. The human newborn therefore is totally dependent on the activity of the innate immune system, as well as on transplacentally obtained antibodies from mother. Timely vaccination is required, because maternal antibodies will have disappeared by 3-6 months. Since more than 50 years, national childhood vaccination schedules have been implemented in most countries. In the Netherlands this program is offered free of charge and on a voluntary basis [15]. Next to young children, several other risk groups are eligible for specific vaccinations. Elderly (> 65 years) and patients with chronic conditions are vaccinated against seasonal influenza and pneumococcal infections. Additionally, for immunocompromised patients who have undergone splenectomy and for patients on immunosuppressive medication, specific vaccines are recommended.. Due to the fact that the immune system of this category of patients does not function optimally, vaccination can be less effective. On the other hand, these immunocompromised individuals, due to their impaired immune response, are especially sensitive to these infectious diseases with potentially severe consequences. Therefore, vaccination of this specific group is highly recommended [16]. For a number of occupations, namely those in health care, additional vaccinations (hepatitis B, influenza vaccination) are strongly recommended if not required. Finally, and in the context of this paper, most importantly, travelers, especially those going to certain risk areas, are eligible for additional, specific vacations [17].
4.The Antibody Response to Vaccination Can be Used as A Correlate of Protection
As mentioned above, the effectiveness of a vaccine is determined by the level of protection conferred against the target disease in the target population. Prior to the introduction of a new vaccine, effectiveness (and safety) needs to be demonstrated in field studies. For individual patients or small groups, the immune response to the vaccine can be used as a correlate for protection. In practice, the serum antibody level is used for this purpose, even in case of viral vaccines. Depending on the vaccine, the vaccination schedule consists either of a single injection or a series of injections. It should be kept in mind that a baby born from a vaccinated mother will have received IgG antibodies from mother [18]. While these transplacentally acquired antibodies will help to protect the infant from infection, these maternal antibodies may inhibit the active immune response of infants to the vaccines [19]. Any vaccination results in a progressively increasing antibody titer, which gradually decreases afterwards. The speed with which the antibody titer decreases after the last injection at the age of 9 is evidently slower than in newborns. Namely, at the age of 15, the titer is still well above the level needed to be considered protected. At the age of 50, more than 95% of the vaccinated individuals still have sufficiently high antibody levels to be protected against poliomyelitis [20].
The substantially gradual decline of the antibody titer after being fully vaccinated cannot be explained based on the halftime of IgG. There are, however, at least three other (theoretical) reasons that could possible explain why the antibody titers decrease so slowly. First of all, there are many long-lasting plasma cells in the bone marrow that can continue to produce antibodies for years [21]. In healthy elderly, for example, IgA and IgG levels of anti- pneumococcal antibodies is known to increase significantly after the age of 70 [22]. Secondly, a small amount of vaccine antigen can persevere. Even though it is being discussed whether this is the primary effect of the adjuvants, the aluminum salts (alum) that are added to vaccines can result in a depot formation [23]. This allows a slow release of antigens to occur over an elongated period of time, causing long-lasting stimulation of the immune system to produce antibodies over a bigger period [24].
5. Polysaccharides Conjugate Vaccines Can Prevent Invasive Bacterial Infections in Infants
Young children, below the age of 5 years, are susceptible to infestations with encapsulated bacteria. These include pneumococci, Haemophilus influenzae type b and meningococci [25]. The capsule of these bacteria consists of polysaccharides, which are the main target of the humoral immune system. Polysaccharides are molecules with a high molecular weight and they consist of 50-150 repeating units of 2 to 4 different sugar molecules which are linear or limited branched. Polysaccharide antigens belong to the category of type 2 T cell-independent antigens because they can induce antibody production without the aid of T lymphocytes [26, 27]. With respect to vaccination, there are 2 other properties of polysaccharides that are important to consider. The first is that polysaccharides induce little, if any, immunological memory, meaning that revaccination (or infection) will not lead to a better response [28]. The second aspect has to do with the onset of responsiveness of the immune system. Responsiveness to polysaccharides in humans develops not before the age of 1.5-2 years. The likely mechanism for the unresponsiveness to polysaccharide early in life is based on the reduced expression of complement receptor type 2 on marginal-zone B cells [29, 30]. Furthermore, the quantitative defect to produce IgG2 immunoglobulins, which is the most effective IgG subclass against (some) polysaccharides, also takes several years to mature [31].
In the 1930’s, Avery and Goebel discovered that the immunogenicity of polysaccharide antigens could be improved by coupling them to a protein carrier (a mixture of polysaccharides and proteins is not sufficient, a physical link is required) [32]. It took approximately 50 years before this principle was rediscovered and applied to improve the Hib polysaccharide vaccine. The coupling of the (Hib) polysaccharide to a protein (e.g. tetanus toxoid) results in a so-called conjugate vaccine which is capable of stimulating anti-Hib polysaccharide antibody production in young children [33]. Because of the coupling to the protein, the Hib polysaccharide response has obtains a T-cell dependent character. A study in 1996 by Zepp and colleagues showed that not only is the conjugate vaccine more immunogenic, but it also has the capability of inducing immunological memory [34]. Most importantly, this conjugate vaccine has the ability of preventing meningitis (and other forms of invasive Hib) in infants in their first year of life. The Hib conjugate vaccine was the first polysaccharide-protein conjugate vaccine to become implemented. Before introduction of Hib vaccination in The Netherlands, annually 700 children under the age of 5 developed invasive Hib disease. Since its introduction in 1993, Hib meningitis has practically disappeared in the Netherlands [35] as in other countries that have implemented Hib conjugate vaccination. The same principle of conjugation with proteins is also applied in vaccines against meningococcus and pneumococcus. The menC conjugate vaccine was introduced in 2002, nowadays at tetravalent meningococcal conjugate vaccine (including the serotypes A, C, W, and Y) is available and being used [36]. The first generation pneumococcal conjugate vaccines consisted of the polysaccharides of 7 of the most prevalent pneumococcal serotypes (4, 6B, 9V, 14, 18C, 19F and 23F). This vaccine protected against approximately 60-70% of the invasive pneumococcal infections (meningitis, bacteremia) in this age group [37]. The effectiveness against mucosal infections such as otitis media is limited. The 7 valent pneumococcal conjugate vaccine has been followed up by 10- and 13-valent pneumococcal conjugates vaccines, in order to address serotype replacement diseases as well expanding coverage [38]. Next generation pneumococcal conjugates probably will be 20-valent.
For travelers, the WHO advises to keep in mind that the composition of the conjugate vaccines are based on seroprevalence in the USA and other industrialized countries. Travelers planning on going to Africa and Asia should therefore take into consideration that the coverage of the vaccines they have had is substantially lower in countries there. During the period of 2001-2013, out of the top-10 serotypes causing pneumonia in Bangladeshi children, only 3 serotypes were included in PCV7 [39]. What is most striking is that the 2 predominant serotypes, 26% combined, are not covered by the PCV7 vaccine and only the second most prevalent one, serotype 1 (10%), is covered by PCV10 and PCV13. This means that travelers, especially children, from the North America and Europe still run the risk of falling ill with severe pneumonia. Serotype 2 is not covered by any of the current PCV vaccines as it had not been detected for the past decades until a sudden upsurge from 2001 onwards in Asia [40]. This could have been due to replacement disease after the introduction of PCV7, as other non-vaccine serotypes have been known to increase in frequency once vaccine-targeted serotypes decrease [41]. However, studies in the UK, the Netherlands and the US have not found serotype 2 as a replacement serotype [42-44]. This underscores the conclusion that global introduction of a pneumococcal conjugate vaccine can lead to replacement disease with different serotypes throughout the world.
6. Adjuvants Can Augment The Effect of Vaccines Through Depot Formation or Facilitation of The Second Lymphocyte Activation Signal
Almost a 100 years ago it was discovered that the antibody response can be greatly enhanced by the addition of certain substances, such as alum, to the antigen preparation. These substances are called adjuvants. Nowadays, alum (especially aluminum hydroxide) is still used, for example in the DTP vaccine.
Adjuvants works in at least two different ways. First of all, they have the ability to concentrate the antigen (the vaccine) on one site, which provides optimal uptake of the antigen by antigen-presenting cells (APCs) and subsequent activation of the immune system [45]. As previously mentioned, alum additives cause prolonged effects; they lead to the formation of small depots which retain the antigen for a long time. Secondly, adjuvants can optimize the second signal needed for activation of the immune response. Adjuvants result in an increased expression of costimulatory molecules and in increase in cytokine secretion [46]. This occurs primarily by means of activation of Toll- like receptors on antigen presenting cells. An example of such an adjuvant is the MPL (monophosphoryl lipid A) that can bind to TLR4 [47].
7. Vaccination Against Every Infectious Disease Is Not (Yet) Possible
Unfortunately, the list of severe infectious diseases for which there are no vaccines or no adequate working vaccines available remains long. High on this list are HIV, tuberculosis (TB) and malaria. In 1984, when it was discovered that the HIV virus was the cause of AIDS, 1300 people had died from this disease. At that time, it was speculated that an effective vaccine for AIDS would become available within 2 years. Now, almost 35 years and more than 35 million casualties later [48], despite enormous efforts, there is still no effective AIDS vaccine. The global HIV/AIDS epidemic has affected no less than 186 countries, which had all reported cases or deaths in 2012 [49]. An indirect consequence of the AIDS epidemic is a strong increase in the incidence of TB. CD4+ T cells have a central role in protecting the body against TB infection [50]. HIV breaks down the human cellular immune system by infecting and killing CD4+ T cells [51]. What causes the high susceptibility of HIV infected people to TB is thus for a large part due to this destruction of the CD4+ T cells, and so the cellular immune response, which should protect the body against TB [52]. Approximately one- third of the world population is infected with M. tuberculosis with 10.4 million new cases of TB in 2016 and 1.7 million deaths (WHO) [53]. The original tuberculosis vaccine (BCG) from 1921 provides insufficient protection (possibly because the vaccine contains Mycobacterium bovis and not Mycobacterium tuberculosis) and therefore there is urgent need for a better vaccine [54,55]. For a large number of parasitic diseases (tropical protozoa and worm infections), there also are no effective vaccines available. In part, this is due to the fact that for parasitic vaccines, knowledge on the mechanisms of the protective immunity is still inadequate. A reason for this is the severely dynamic antigenicity of these organisms. Another limiting factor is that the most important parasitic infections (malaria, Schistosoma, Leishmania) are especially prevalent in the tropics and developmental countries [56]. Therefore, there is not a high economic incentive for the pharmaceutical industry to invest in the development of such vaccines. Thus, the road to an effective malaria vaccine is long and winding [57]. An experimental malaria vaccine appears to indicate signs of clinical protection against Plasmodium falciparum infection [58]. This so-called RTS, S vaccine consists of a fusion-protein of the P. falciparum circumsporozoite protein and hepatitis B surface antigen (hBsAg). The vaccine is used with an adjuvant mixture consisting of bacterial cell wall components and a plant-based component (AS02D). This vaccine has been tested as safe and immunogenic in infants and young children with a 66% protection against malaria [59]. The most recent developments in this field hold promise for development of effective malaria vaccines, either passive or active [60-62].
The above mentioned vaccination approaches are all committed aimed at preventing infection. Vaccination can also be used to prevent (viral induced) cancers. Currently, vaccination for cervical cancer prevention, caused by papilloma viruses has been implemented [63]. Human papillomaviruses (HPV) form a variable group of approximately a 100 different DNA viruses that can infect the skin and mucosal linings. A large number of HPV types are spread via skin-to-skin contact, and a smaller group of about 13 types are spread through sexual contact. The latter can cause genital malignancies, especially cervical cancer [64]. Within this group, HPV-16 and HPV-18 are responsible for 70% of cases of cervical cancer. Worldwide there are 500,000 new cases of cervical carcinoma annually, with half of these cases being fatal [65].
Effective defense against HPV relies on both the humoral and the cellular immune response [66]. In 50% of the normal, healthy population, HPV specific CD4+helper T lymphocytes are detectable, indicative for previous contact with HPV and a response of the immune system [67]. However, in the majority of patients with diagnosed HPV induced cervical cancer, there is apparent reduction if not total lack of HPV specific CD4+helper T lymphocytes. Currently, two HPV vaccines have been developed and brought to the market, a bivalent vaccine with HPV-16 and -18 antigens and a quadrivalent vaccine that includes the previous two as well as HPV-6 and -11 (for genital warts) [68]. In the bivalent vaccine, both aluminum hydroxides and monophosphoryl lipid A are added as adjuvants. These vaccines provided young women who had no prior contact with HPV an almost 100% protection against vaccine types [69]. The duration of the protection remains to be established, but studies from 2014 have found a 100% efficacy for short-term (≤5 years), a high efficacy for long-term (≥ 5 years, in this case 8 years) and even seropositivity up to 9 years after vaccination [70, 71]. The protection is less when an individual has already come into contact with one of the HPV types and the vaccine does not have any effect on existing tumors.
8. The Development of A New Vaccine Takes Time
The development of a new vaccine on average, from basic research up until registration, takes 10 years or more. The diseases for which no vaccine has been found (such as HIV) is not included in this figure. It took Calmette and Guerin 13 years to develop the BCG vaccine [72], while the vaccine against to new influenza A (N1H1) was marketed within half a year.
When a new vaccine is brought onto the market, it will take a certain period before it is generally accepted to use the vaccine (and to possible add it to national vaccination schedules). After the introduction of a successful vaccine, the disease burden will decrease to such an extent that the general population, and even doctors, will not recognize the disease in question anymore. When a vaccination coverage of 95% is achieved, the small minority of the population that has not been vaccinated or for whom the vaccine does not work, is protected by herd immunity. A decline in the coverage may however result in a new outbreak of the infectious disease. For example, the risk of a measles epidemic increases enormously when the immunization coverage drops below 75%. Ideally, mass vaccination will lead to complete eradication of a disease, as with smallpox. In that case, vaccination will not be necessary anymore and can be stopped. In the meantime, and in light of the long list of candidate vaccines, national vaccination schedules will not reduce but expand. The risk, sometimes ventilated, that the immune system of young children will be overloaded with all these vaccines is not real. As compared to all other daily antigenic stimuli for the immune system, vaccines are just a minor fraction [73].
9. Vaccination For Immuno Competent and Immuno compromised Travelers
Travel vaccination is recommended for all persons who travel to areas where certain infectious diseases may be prevalent because of climatological and/or hygienic circumstances. It is remarkable that the great discoverers of the past, who explored the world and reached places where they will have been exposed to a whole array of potential pathogens, hardly would have benefited from travelers vaccination (Figure 3).
With an increasing number of vaccines available, there is no universal applicable vaccination schedule for all travelers to any destination. Minimally required are the vaccines from the national vaccination programs. While this may sound obvious, it isn’t. In the Netherlands the national vaccination program was implemented 60 years ago, so older people may not have been fully vaccinated. When living in an environment with a high vaccination rate, the risk for infection of non-vaccinated individuals is low due to herd immunity. When traveling to an area with a low vaccination rate, herd immunity will be absent and the risk for exposition and infection will be substantially higher. Which other vaccines will be required or recommended depends on the specific destination, the duration of the stay, and additional risk factors and/or underlying diseases (see also below). A complete advice for travelers will not be restricted to vaccination but will also include information on additional preventive measures against malaria and other infectious diseases for which no (effective) vaccines or adequate treatment options are available.
Most vaccinations for travelers are given on a voluntary basis, some however are compulsory. An example of compulsory vaccination is yellow fever when traveling to areas where this disease is endemic (roughly the jungle covered areas of Africa and South America). Sometimes neighboring countries demand travelers to be vaccinated in order to prevent spread of the virus. Meningococcal vaccination (A,C,W, and Y) is compulsory for pilgrims to Mecca and other holy places in Saudi Arabia.
Immunocompromised patients are at increased risk of infections, a number of which can be prevented by vaccination [74]. These patients however also are less likely to mount an effective immune response leading to the clinical paradox that precisely the group that would need most protection is least likely to respond adequately to vaccination [75]. It should be added that the rate of exposure or even infection needs to be higher in immunocompromised travelers, but the illnesses will have significantly more severe consequences [76]. The same vaccination rules apply to the immunocompromised traveler as to someone with a fully functional immune system. However, there are a few additional things to keep in mind. These travelers should seek pre- travel consultations 4-6 weeks ahead of their travel according to the WHO. This should give the traveler plenty of time to update their immunization status, consult infectious diseases specialists and primary physicians as well as any other specialist relevant to the reason behind the traveler’s immunocompromised state if needed. In a study from 2002 until 2012, a Swiss travel clinic collected data of the travel patterns of their clients as well pre- and post-travel consultation. A total of 65,046 clients were included of which 30.8% self-indicated they had a type of immunosuppressive condition (cancer, HIV, splenectomy, etc.) [77]. A small number of them (0.1%) was taking immunosuppressive medication (prednisone, methotrexate, etc.). Africa and Asia were the most visited continents, 46% and 35% respectively, with India being the most frequently visited country (9.6%). Less than half of the travelers had consulted medical professionals about their travel plans 1 month or more prior to their departure date and similar data were found for Swedish travelers [78]. However, while most travelers did not seek pre-travel advice at the right time, the study indicated that 99% did in fact receive their required vaccines. In the USA, a comparable study of 13,235 travelers from 2001 until 2011 was conducted [79] As in Europe, the most frequently visited continent and country were Africa and India. The pre-travel advice period was 14-17 days for US travelers. While there were more travelers with pre-existing medical conditions (59% USA, 46.5% Switzerland), only 3% of the American travelers had an immunocompromising condition. In both studies it was reported that some travelers declined vaccination or were advised against certain vaccines. The US Centers for Disease Control and Prevention (CDC) have defined different categories of an immunocompromised state [80]. Based on this classification, it is easier to determine which vaccines are or are not recommended for a certain category of immunocompromised travelers [81].
Immunocompromised patients are a growing group within the population. With the continuous development of novel immunosuppressive medication, as well as improved survival of various groups of patients,many patients generally feel healthy and physically and mentally fit to embark on international travels, putting them at risk of (serious) infections.
10.Conclusion
Vaccination against infectious diseases is the most clear-cut example and large-scale application of immunology in practice. Vaccination has largely contributed to the improvement of overall health and increase in the life-expectancy in the last 50 years. In case a new disease breaks out, such as Ebola, Zika virus and Q fever, the demand for novel vaccines will arise. National vaccination schedules are aimed at the most prevalent infectious diseases for the respective country or region. (International) travelers will need additional vaccination, depending on their destination. Over the past decade, the treatment of patients with immune mediated diseases and with tumors has improved dramatically. Most of the biologicals used for these treatments have an impact on the functioning of the immune system. As a consequence, vaccines may be less effective. This has consequences for immune compromised travelers, because standard dosing of vaccines may not be sufficient.
References
- Calendarium Romanum. https://archive.org/ details/CalendariumRomanum1969 Assessed April 14, 2018
- Shah MK. The Immunocompromised Traveler. Oncology (Williston Park) 2016; 30(2): 142- 146.
- Medzhitov R. Origin and physiological roles of inflammation. Nature 2008; 454(7203):428.
- Vilella A, Bayas JM, Diaz MT, Guinovart C, Diez C, Simó D, Muñoz A, Cerezo J. The role of mobile phones in improving vaccination rates in travelers. Preventive medicine 2004;38(4):503- 9.
- Stern AM, Markel H. The history of vaccines and immunization: familiar patterns, new challenges. Health Affairs. 2005; 24(3):611-21.
- Riedel S. Edward Jenner and the history of smallpox and vaccination. Proc (Bayl Univ Med Cent) 2005; 18(1):21-25.
- Keller MA, Stiehm ER. Passive immunity in prevention and treatment of infectious diseases. Clinical microbiology reviews. 2000;13(4):602- 14.
- Hansson GK, Libby P, Schönbeck U, Yan ZQ. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circulation research. 2002;91(4):281-91.
- Jäger E, Chen YT, Drijfhout JW, Karbach J, et al. Simultaneous humoral and cellular immune response against cancer–testis antigen NY-ESO- 1: Definition of human histocompatibility leukocyte antigen (HLA)-A2–binding peptide epitopes. Journal of Experimental Medicine. 1998;187(2):265-70.
- Mahanty S, Prigent A, Garraud O. Immunogenicity of infectious pathogens an d vaccine antigens. BMC immunology. 2015;16(1):31.
- Plotkin S.A., and Gilbert, P.B. Nomenclature for immune correlates of protection after vaccination. Clin Infect Dis. 2012; 54(11):1615- 7.
- Pirofski LA, Casadevall A. Use of licensed vaccines for active immunization of the immunocompromised host. Clinical microbiology reviews. 1998; 11(1):1-26.
- Espeland EM, Tsai CW, Larsen J, Disbrow GL. Safeguarding against Ebola: Vaccines and therapeutics to be stockpiled for future outbreaks. PLoS Negl Trop Dis. 2018; 12(4):e0006275.
- PrabhuDas M, Adkins B, Gans H, King C, Levy O, Ramilo O, Siegrist CA. Challenges in infant immunity: implications for responses to infection and vaccines. Nature immunology. 2011; 12(3):189.
- Van Lier EA, Oomen PJ, Giesbers H, Conyn- van Spaendonck MA, et al. Vaccinatiegraad Rijksvaccinatieprogramma Nederland: Verslagjaar 2014.
- Kunisaki KM, Janoff EN. Influenza in immunosuppressed populations: a review of infection frequency, morbidity, mortality, and vaccine responses. The Lancet infectious diseases. 2009;9(8):493-504.
- Zuckerman JN. Travelers' Vaccines. PMPH- USA; 2010.
- Morell A, Skvaril F, Hitzig WU, Barandun S. IgG subclasses: development of the serum concentrations in “normal” infants and children. The Journal of pediatrics. 1972; 80(6):960-4.
- Jones C, Pollock L, Barnett SM, Battersby A, Kampmann B. The relationship between concentration of specific antibody at birth and subsequent response to primary immunization. Vaccine. 2014;32(8):996-1002.
- Conyn-Van Spaendonck MA, de Melker HE, Abbink F, Elzinga-Gholizadea N, Kimman TG, van Loon T. Immunity to poliomyelitis in The Netherlands. Am J Epidemiol. 2001; 153(3): 207-14.
- Kometani K, Kurosaki T. Differentiation and maintenance of long-lived plasma cells. Current opinion in immunology. 2015; 33:64-9.
- Bátory G, Jancsó Á, Puskás É, Rédei A, Lengyel É. Antibody and immunoglobulin levels in aged humans. Archives of gerontology and geriatrics. 1984; 3(2):175-88.
- Coffman RL, Sher A, Seder RA. Vaccine adjuvants: putting innate immunity to work. Immunity. 2010;33(4):492-503.
- Lambrecht BN, Kool M, Willart MA, Hammad H. Mechanism of action of clinically approved adjuvants. Current opinion in immunology. 2009; 21(1):23-9.
- Watt JP, Wolfson LJ, O'Brien KL, Henkle E, et al. Burden of disease caused by Haemophilus influenzae type b in children younger than 5 years: global estimates. The Lancet. 2009; 374(9693):903-11.
- Pollard AJ, Perrett KP, Beverley PC. Maintaining protection against invasive bacteria with protein–polysaccharide conjugate vaccines. Nature Reviews Immunology. 2009; 9(3):213.
- Rijkers G. Cutting the Stone: Health Defined in the Era of Value-based Care. Cureus. 2017; 9(2):e1023.
- de Roux A, Schmöele-Thoma B, Siber GR, Hackell JG, et al. Comparison of pneumococcal conjugate polysaccharide and free polysaccharide vaccines in elderly adults: conjugate vaccine elicits improved antibacterial immune responses and immunological memory. Clinical Infectious Diseases. 2008; 46(7):1015- 23.
- Klein Klouwenberg P, Bont L. Neonatal and infantile immune responses to encapsulated bacteria and conjugate vaccines. Clinical and Developmental Immunology. 2008 Sep 23;2008.
- Griffioen AW, Franklin SW, Zegers BJ, Rijkers GT. Expression and functional characteristics of the complement receptor type 2 on adult and neonatal B lymphocytes. Clin Immunol Immunopathol. 1993;69(1):1-8.
- Barrett DJ, Ayoub EM. IgG2 subclass restriction of antibody to pneumococcal polysaccharides. Clinical and experimental immunology. 1986;63(1):127.
- Avery O, Goebel WF. Chemo-immunological Studies on Conjugated Carbohydrate-proteins. II. Immunological Specificity of Synthetic Sugar protein Antigens. J Exp Med 1929; 50:533.
- Peltola H. Worldwide Haemophilus influenzae type b disease at the beginning of the 21st century: global analysis of the disease burden 25 years after the use of the polysaccharide vaccine and a decade after the advent of conjugates. Clinical microbiology reviews. 2000; 13(2):302- 17.
- Zepp F, Schmitt HJ, Kaufhold A, Schuind A, Knuf M, Habermehl P, Meyer C, Bogaerts H, Slaoui M, Clemens R. Evidence for induction of polysaccharide specific B-cell-memory in the 1st year of life: plain Haemophilus influenzae type b-PRP (Hib) boosters children primed with a tetanus-conjugate Hib-DTPa-HBV combined vaccine. European journal of pediatrics. 1996; 156(1):18-24.
- Monge S, Mollema L, de Melker H, Sanders E, van der Ende A, Knol M. Clinical Characterization of Invasive Disease Caused by Haemophilus influenzae Serotype b in a High Vaccination Coverage Setting. J Pediatric Infect Dis Soc. 2018. doi: 10.1093/jpids/piy020.
- Merino Arribas JM, Carmona Martínez A, Horn M, et al. Safety and Immunogenicity of the Quadrivalent Meningococcal Serogroups A, C, W and Y Tetanus Toxoid Conjugate Vaccine Coadministered With Routine Childhood Vaccines in European Infants: An Open, Randomized Trial. Pediatr Infect Dis J. 2017; 36(4):e98-e 107.
- Whitney CG, Farley MM, Hadler J, et al. Decline in invasive pneumococcal disease after the introduction of protein-polysaccharide conjugate vaccine. N Engl J Med. 2003; 348(18):1737-46.
- Rijkers GT, Yousif LI, Spoorenberg SM, van Overveld FJ. Triptych of the Hermit Saints: pneumococcal polysaccharide vaccines for the elderly. Risk Manag Healthcare Policy. 2018; 11:55-65.
- Saha SK, Hossain B, Islam M, Hasanuzzaman M, et al. Epidemiology of invasive pneumococcal disease in Bangladeshi children before introduction of pneumococcal conjugate vaccine. The Pediatric infectious disease journal. 2016; 35(6):655-61.
- Saha SK, Al Emran HM, Hossain B, Darmstadt GL, Saha S, et al. Streptococcus pneumoniae serotype-2 childhood meningitis in Bangladesh: a newly recognized pneumococcal infection threat. PLoS One. 2012; 7(3):e32134.
- Fitzwater SP, Chandran A, Santosham M, Johnson HL. The worldwide impact of the seven-valent pneumococcal conjugate vaccine. Pediatr Infect Dis J. 2012;31(5):501-8.
- Miller E, Andrews NJ, Waight PA, Slack MP, George RC. Herd immunity and serotype replacement 4 years after seven-valent pneumococcal conjugate vaccination in England and Wales: an observational cohort study. Lancet Infect Dis. 2011;11(10):760-8.
- Spijkerman J, van Gils EJ, Veenhoven RH, Hak E, et al. Carriage of Streptococcus pneumoniae 3 years after start of vaccination program, the Netherlands. Emerging infectious diseases. 2011; 17(4):584.
- Huang SS, Platt R, Rifas -Shiman SL, Pelton SI, Goldmann D, Finkelstein JA. Post-PCV7 changes in colonizing pneumococcal serotypes in 16 Massachusetts communities, 2001 and 2004. Pediatrics. 2005;116(3):e 408-13.
- Rock KL, Hearn A, Chen CJ, Shi Y. Natural endogenous adjuvants. InSpringer seminars in immunopathology 2005 Jan 1 (Vol. 26, No. 3, pp. 231-246). Springer-Verlag.
- Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453(7198):1122.
- Evans JT, Cluff CW, Johnson DA, Lacy MJ, Persing DH, Baldridge JR. Enhancement of antigen-specific immunity via the TLR4 ligands MPL adjuvant and Ribi.529. Expert Rev Vaccines. 2003; 2(2):219-29.
- World Health Organization Global Health Observatory Data HIV/AIDS http://www.who. int/gho/hiv/en/ accessed April 9 2018
- Ortblad KF, Lozano R, Murray CJ. The burden of HIV: insights from the Global Burden of Disease Study 2010. AIDS (London, England). 2013;27(13):2003.
- Cooper AM. Cell-mediated immune responses in tuberculosis. Annual review of immunology. 2009; 27:393-422.
- Alimonti JB, Ball TB, Fowke KR. Mechanisms of CD4+ T lymphocyte cell death in human immunodeficiency virus infection and AIDS. Journal of general Virology. 2003; 84(7):1649- 61.
- Tufariello JM, Chan J, Flynn JL. Latent tuberculosis: mechanisms of host and bacillus that contribute to persistent infection. The Lancet infectious diseases. 2003;3(9):578-90.
- Global tuberculosis report 2017. Geneva: World Health Organization; 2017. Licence: CC BY- NCSA 3.0 IGO. https://creativecommons.org/ licenses/by-nc-sa/3.0/igo accessed April 9 2018
- Mahairas GG, Sabo PJ, Hickey MJ, Singh DC, Stover CK. Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M. bovis. Journal of bacteriology. 1996; 178(5):1274-82.
- World Health Organization. BCG vaccine: WHO position paper, February 2018 - Recommendations. Vaccine. 2018. pii: S0264- 410X(18)30345-1. doi:10.1016/ j.vaccine.2018. 03.009.
- Greenwood B, Mutabingwa T. Malaria in 2002. Nature. 2002 ;415(6872):670.
- Daar AS, Thorsteinsdóttir H, Martin DK, Smith AC, Nast S, Singer PA. Top ten biotechnologies for improving health in developing countries. Nature genetics. 2002;32(2):229.
- Bojang KA, Milligan PJ, Pinder M, Vigneron L, Alloueche A, et al. Efficacy of RTS, S/AS02 malaria vaccine against Plasmodium falciparum infection in semi-immune adult men in The Gambia: a randomised trial. The Lancet. 2001; 358(9297):1927-34.
- Casares S, Brumeanu TD, Richie TL. The RTS, S malaria vaccine. Vaccine. 2010;28(31):4880- 94.
- Kisalu NK, Idris AH, Weidle C, Flores -Garcia Y, et al. A human monoclonal antibody prevents malaria infection by targeting a new site of vulnerability on the parasite. Nat Med. 2018 Mar 19. doi: 10.1038/nm.4512.
- Sack BK, Mikolajczak SA, Fishbaugher M, Vaughan AM, et al. Humoral protection against mosquito bite-transmitted Plasmodium falciparum infection in humanized mice. NPJ Vaccines. 2017; 2:27. doi: 10.1038/s41541-017- 0028-2.
- Zenklusen I, Jongo S, Abdulla S, Ramadhani K, et al. Immunization of malaria pre-exposed volunteers with PfSPZ Vaccine elicits long - lived IgM invasion-inhibitory and complement- fixing antibodies. J Infect Dis. 2018. doi: 10.1093/infdis/jiy080.
- Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nature Reviews Immunology. 2005;5(4):296.
- Winer RL, Lee SK, Hughes JP, Adam DE, Kiviat NB, Koutsky LA. Genital human papillomavirus infection: incidence and risk factors in a cohort of female university students. American journal of epidemiology. 2003; 157(3):218-26.
- Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. International journal of cancer. 2010; 127(12):2893-917.
- Stanley M. Immune responses to human papillomavirus. Vaccine. 2006 ;24:S16-22.
- Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40–CD40L interactions. Nature. 1998;393(6684):480.
- Jit M, Chapman R, Hughes O, Choi YH. Comparing bivalent and quadrivalent human papillomavirus vaccines: economic evaluation based on transmission model. BMJ. 2011; 343:d5775.
- Harper DM, Franco EL, Wheeler CM, Moscicki AB, et al, HPV Vaccine Study group. Sustained efficacy up to 4• 5 years of a bivalent L1 virus - like particle vaccine against human papillomavirus types 16 and 18: follow-up from a randomised control trial. The Lancet. 2006;367(9518):1247-55.
- Deleré Y, Wichmann O, Klug SJ, van der Sande M, Terhardt M, Zepp F, Harder T. The efficacy and duration of vaccine protection against human papillomavirus: a systematic review and meta-analysis. Deutsches Ärzteblatt International. 2014;111(35-36):584.
- De Vincenzo R, Conte C, Ricci C, Scambia G, Capelli G. Long-term efficacy and safety of human papillomavirus vaccination. International journal of women's health. 2014;6:999.
- Barberis I, Bragazzi NL, Galluzzo L, Martini M. The history of tuberculosis: from the first historical records to the isolation of Koch 's bacillus. J Prev Med Hyg. 2017; 58(1):E9-E12.
- Hulsey E, Bland T. Immune overload: Parental attitudes toward combination and single antigen vaccines. Vaccine. 2015; 33(22):2546-50.
- Backhaus E, Berg S, Andersson R, et al. Epidemiology of invasive pneumococcal infections: manifestations, incidence and case fatality rate correlated to age, gender and risk factors. BMC Infect Dis 2016; 16: 367.
- Lobermann M, Borso D, Hilgendorf I, Fritzsche C, Zettl UK, Reisinger EC. Immunization in the adult immunocompromised host. Autoimmun Rev 2012; 11(3): 212-8.
- Baaten GG, Geskus RR, Kint JA, Rouken AH, Sonder GJ, van den Hoek A. Symptoms of infectious diseases in immunocompromised travelers: a prospective study with matched controls. J Travel Med. 2011; 18(5):318–326.
- Boubaker R, Meige P, Mialet C, Ngarambe Buffat C, et al. Travellers’ profile, travel patterns and vaccine practices —a 10-year prospective study in a Swiss Travel Clinic. Journal of travel medicine. 2016; 23(1).
- Dahlgren AL, Deroo L, Steffen R. Prevention of travel-related infectious diseases: knowledge, practices and attitudes of Swedish travellers. Scandinavian journal of infectious diseases. 2006;38(11-12):1074-80.
- LaRocque RC, Rao SR, Lee J, Ansdell V, Yates JA, et al. Global TravEpiNet: a national consortium of clinics providing care to international travelers —analysis of demographic characteristics, travel destinations, and pretravel healthcare of high-risk US international travelers, 2009–2011. Clinical infectious diseases. 2011;54(4):455-62.
- Centers for Disease Control and Prevention (CDC) CDC Health Information for International Travel 2014. New York: Oxford University Press; 2014.
- Rubin LG, Levin MJ, Ljungman P, Davies EG, Avery R, et al. 2013 IDSA clinical practice guideline for vaccination of the immunocompromised host. Clinical Infectious Diseases. 2013; 58(3):e44-100.