


|
|
DOI Prefix 10.20431 |
Information
Journal Policies
Adult Onset Progressive Symmetric Erythrokeratoderma: A Case Report
Deepthi Ravi1,Keerthana Reddy2,Jayakar Thomas3*
2.Junior Residents, Department of Dermatology, Venereology & Leprosy, Sree Balaji Medical College & Hospital, Bharath University, Chennai 600044, Tamil Nadu, India.
3.HOD & Professor, Department of Dermatology, Venereology & Leprosy, Sree Balaji Medical College & Hospital, Bharath University, Chennai 600044, Tamil Nadu, India.
Copyright : © 2018 Authors. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Progressive symmetric erythrokeratoderma (PSEK) is a rare genodermatoses usually having an autosomal dominant mode of inheritance but other modes such as autosomal recessive have also been reported [1]. Patients usually present with symmetrical erythematous scaly plaques over the extremities and the trunk. We report a case of PSEK in a 46 year old male patient born of a second-degree consanguineous marriage.
Keywords: Progressive symmetric erythrokeratoderma, erythematous plaques, genodermatoses, Dermatology
1. Introduction
Erythrokeratodermas are a rare genodermatoses and are usually inherited in an autosomal dominant manner with incomplete penetrance and variable expression [2]. There are two main non-syndromic types of erythrokeratodermas namely progressive symmetric erythrokeratoderma (PSEK) also known as Gottron’s syndrome and erythrokeratoderma variabilis.
PSEK was described in 1886 by Darier and Gottron gave the terminology of progressive symmetrical erythrokeratoderma in 1922. PSEK usually has a childhood onset and has equal gender incidence.
It is proposed to occur due to a mutation in the loricrin protein which has a gene coding on 1q21.3. Loricin is a major component of the cornified cell envelope epithelium which is formed during terminal differentiation below the plasma membrane of stratified squamous epithelial cells.
We report a case of adult onset PSEK in a 46 year old male patient with possible autosomal recessive inheritance.
2. Case Report
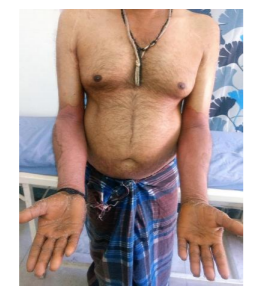
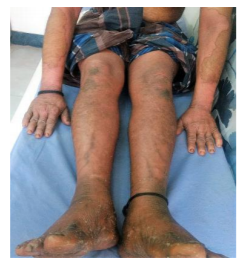
A 46 year old male patient born of a second degree consanguineous (father married his sister’s daughter) marriage presented with complaints of erythematous skin lesions over the body since the last 6 months. It started over the right arm and progressed to involve the abdomen, legs, elbows, knees, palms and soles. It started as a raised erythematous lesion which subsequently developed scaling. Pigmentation developed in a few lesions over the elbows and knees. The patient had been diagnosed as erythrokeratoderma variabilis in the past and was treated with isotretinoin without much improvement. He was later treated with acitretin with slight improvement; however, the patient discontinued treatment.
No family history of similar lesions. No history of any other co-morbidities. History of alcohol consumption and smoking present. Stopped alcohol consumption one year back.
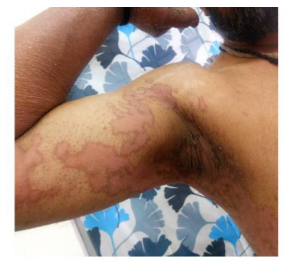
On examination multiple well defined symmetrical erythematous plaques with tiny white pityriasiform scales and a pigmented to erythematous border were seen over the axillae, groins, cubital, popliteal fossae, forearms, arms, legs, thighs, knees and elbows as well as the back and the abdomen. Palmoplantar hyperkeratosis was seen. Pigmented areas were seen on the plaques over the elbows and knees.
Intertrigo was present over the groin crease. Crusting of a few lesions were seen along with a foul odour. There was thickening, subungual hyperkeratosis, distal onycholysis and swelling of the nail folds of the finger and toe nails. Oral cavity, scalp, face was normal.
Bilateral pedal edema was present. Systemic examination was within normal limits. The patient had clinical depression with suicidal tendencies due to his skin condition and was on antidepressive agents.
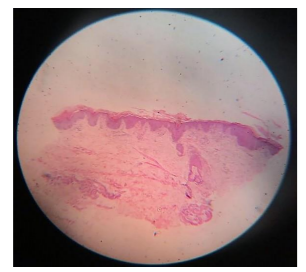
Biopsy was taken from the lesion over the right forearm and showed massive hyperkeratosis, acanthosis, intact granular layer, papillomatosis and lymphocytic infiltration in the papillary dermis.
Based on the clinical and histological findings a diagnosis of progressive symmetric erythrokeratoderma was made and the patient was re-started on acitretin 20mg per day along with systemic antibiotics, topical keratolytics and emollients.
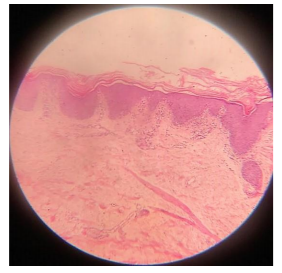
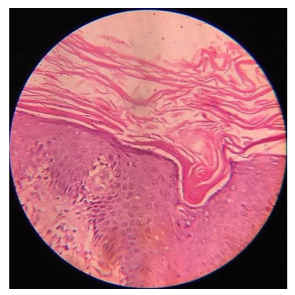
3. Discussion
PSEK usually presents in the first decade of life with well-defined erythematous scaly plaques over the shoulders, gluteal region, extremities and the face. Progression of the disease is usually seen over a few years following which it remains stable. There is marked improvement following puberty. Positive family history is seen in around half of the cases.
The differentials include erythrokeratoderma variabilis (EKV), psoriasis and pityriasis rubra pilaris. The lesions of EKV are generally migratory and fluctuate in their configuration and extent and has a predisposition to the trunk in addition to the extremities. It also shows seasonal variation.
Psoriasis and pityriasis rubra pilaris (PRP) are generally ruled out on histopathology. Psoriasis as hypogranulosis and alternating hyper and parakeratosis in a checker board pattern which is absent in PSEK.
Reports in literature of the conditions associated with PSEK include nephrotic syndrome, fissured tongue, high arched palate[3], symmetric syndactylism[4], keratosis pilaris. Pectus excavatum, convulsions and delayed mile stones.
This case has been reported for its late onset of which only two cases have been reported in literature [5,6].
4. Conclusion
PSEK usually has a childhood onset and an autosomal dominant mode of inheritance. Here we report an adult onset PSEK. Since the patient did not have any family history of the disorder and a history of consanguinity in the parents, it is more suggestive of an autosomal recessive mode of inheritance. Hence even though it is a rare condition, it should be considered as a differential when a patient presents with symmetrical well defined scaly erythematous plaques with a predisposition to the extremities even with an atypical age of presentation and absence of family history.
References
- Thomas J, Begum C Z, Sharada R G. Symmetrical progressive erythrokeratoderma in siblings. Indian J Paediatr Dermatol 2015; 16:243-5
- Yan HB, Zhang J, Liang W, Zhang HY, Liu JY. Progressive symmetric erythrokeratoderma: Report of a Chinese family. Indian J Dermatol Venereol Leprol 2011; 77:597-600
- Gupta S, Kataria U. Symmentrical progressive erythrokeratoderma. Indian J Dermatol Venereol Leprol. 1999; 65:191-2
- Jaiswal AK. Progressive symmetric erythrokeratoderma associated with symmetric syndactylism. Indian J Dermatol Venereol Leprol. 1994; 60:343-4.
- Prabhu S, Shenoi SD, Pai SB, Handattu S, Bhattachan B. Progressive and symmetric erythrokeratoderma of adult onset: A rare case. Indian Dermatol Online J 2010; 1:43-5.
- Erbagci Z, Tuncel AA, Deniz H. Erythrokeratodermia variabilis with adult onset: Report of a sporadic case unresponsive to systemic retinoids. J Dermatol Ther 2006; 17: 187-9