


|
|
DOI Prefix 10.20431 |
Information
Journal Policies
Case of Omenn Syndrome with a Novel RAG2 Missense Mutation
Tomotaka Mabuchi1,Takashi Matsuyama2,Hanako Yamaoka3
Copyright : © 2018 Authors. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Omenn syndrome is one of rare severe immunodeficiency. It is characterized by the presence of a substantial number of oligoclonal, activated T cells, and the lack of B lymphocytes. Patients with Omenn syndrome usually die from opportunistic infections early in life. The cause of Omenn syndrome is mutations of the recombination activating genes (RAG1 or RAG2). Herein, we report a newborn Japanese case of Omenn syndrome with a novel RAG2 missense mutation. Although it is rare, dermatologists should be aware of Omenn syndrome as a cause of erythroderma in early infancy.
Keywords:Omenn syndrome, RAG1, RAG2,Dermatology
1. Introduction
Omenn syndrome, which was first reported by Omenn in 1985[1], is one of rare severe immunodeficiency. It is characterized by the presence of a substantial number of oligoclonal, activated T cells, and the lack of B lymphocytes [2]. Patients with Omenn syndrome present erythroderma, lymphadenopathy, hepatosplen omegaly, and opportunistic infections early in infancy [2]. Thus, it is fatal disease.
2. Case Report
A newborn Japanese boy was consulted with whole body erythema on day 54 of birth. He was delivered by Caesarean section due to cephalopelvic disproportion at 39 weeks gestation. His parents were not in consanguineous marriage. Erythema had begun showing on his face, scalp, and trunk on day 20, rapidly spreading to the whole body. On day 25, he was admitted to pediatric department of Tokai University Hachioji Hospital because of fever and vomiting. In order to receive detailed examination and treatment of hypereosinophilia and immunodeficiency, he was transferred to the pediatric department of Tokai University Hospital on day 53 of birth.
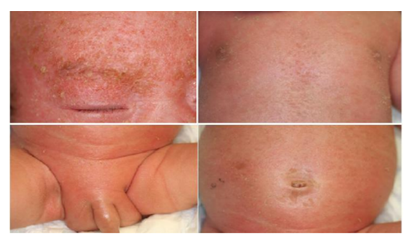
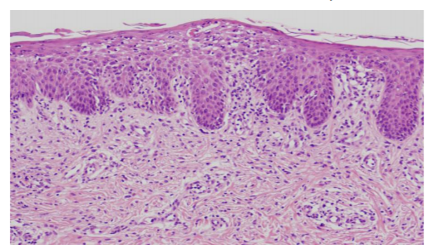
On initial dermatological examination, we noticed whole body erythema with seborrheic scales and crusts (Figure 1). He had neither mucosal lesion nor lymphadenopathy. Nikolsky sign was negative. He had hepatomegaly but not splenomegaly. Laboratory findings were as follows: white blood cell (WBC) count, 19,000 /μL (neutrophils, 44.0 %; lymphocytes, 30.0 %; monocytes, 3.0 %; eosinophils, 17.0 %; and basophils, 0 %); hemoglobin, 8.8 g/dL; platelet, 34.8x104 /μL, reticulocytes, 36 /μL; total protein, 4.6 g/dL; albumin, 2.9 g/dL; creatine phosphokinase (CPK), 29 U/L; aspartate aminotransferase (AST), 129 U/L; aspartate aminotransferase (ALT), 55 U/L; lactate dehydrogenase (LDH), 457 U/L; total bilirubin, 0.5 mg/dL; creatinine, 0.2 mg/dL; blood urea nitrogen (BUN), 9 mg/dL; C-reactive protein (CRP), 2.76 mg/dL; IgG, 57 mg/dL; IgA, < 2 mg/dL; IgM, 2 mg/dL; and IgE, < 1 mg/dL. A skin biopsy was performed from the plaque on his chest. Histopathological examination showed mild spongiosis of epidermis and mild lymphocytic infiltration around vessels in the upper dermis (Figure 2). Immunohistochemistry showed the dermal infiltrate consist of CD3+ T cells.
Differential diagnosis included primary immunodeficiency syndrome such as severe combined immunodeficiency (SCID), Omenn syndrome, and Wiskott-Aldrich syndrome. We consulted Department of Pediatrics, Institute of Medical, Pharmaceutical and Health Sciences, Kanazawa University, Japan to perform genome sequencing. PCR direct sequencing method revealed a novel homozygous mutation of G2530T (Met443Ile) of RAG2 gene. His parents proved to be heterozygous carriers of the same mutation. T cell receptor (TCR)Vβ studies revealed an oligoclonal proliferation of both CD4+ and CD8 + T cells. These findings led to the diagnosis of Omenn syndrome.
He underwent unrelated cord blood stem cell transplantation on day 111 of birth, however, he died 2 days after the transplantation.
3. Discussion
Omenn syndrome is caused by mutations of the recombination activating genes (RAG1 or RAG2)[2]. RAG1 and RAG2 proteins initiate the V(D)J recombination process, which ultimately enables the generation of T cells and B cells with a diversified repertoire of antigen-specific receptors[3]. Mutations in the RAG1 or RAG2 genes are associated with a broad spectrum of clinical phenotypes, ranging from severe combined immunodeficiency to autoimmunity[4]. Typical cases of Omenn syndrome are the result of missense mutation in RAG1 or RAG2 gene and partial recombinase activity[2]. A phenotype of SCID, T-B-NK-SCID bears a defect in either RAG1 or RAG2 gene[2]. Artemis deficiency also causes Omenn syndrome-like phenotype, T-B-NK+ SCID[2].
In this case, genome sequencing revealed a novel homozygous mutation of G2530T (Met443Ile) of RAG2 gene. The core of RAG2 gene spans amino acids 1-387 and the non-core of RAG2 spans amino acids 388-527[3]. Multiple biochemical functions including chromatin and phosphoinositide binding and regulation of protein turnover reside in the non-core of RAG2[3]. The zinc-binding plant homeodomain (PHD) module which spans amino acids 414-487 of non-core of RAG2 is required for normal immune development[3]. Many human patients with primary immune deficiencies such as SCID and Omenn syndrome have been identified with amino acid substitutions in the non-core of RAG2[3]. Our case also had the amino acid substitutions in the non-core of RAG2.
Omenn syndrome is invariably fatal disease. Patients with Omenn syndrome usually die from opportunistic infections early in life[2]. Bone marrow transplantation or cord blood stem cell transplantation is the treatment of choice.
In conclusion, although Omenn syndrome is rare, dermatologists should be aware of Omenn syndrome as a cause of erythroderma in early infancy.
Acknowledgments
We would like to thank Prof. Akihiro Yachie and Dr. Taizo Wada for genome sequencing.
References
- Omenn, G. S. 1965. Familial Reticuloendotheliosis with Eosinophilia. N. Engl. J. Med. 273: 427-432.
- Kato, M., H. Kimura, M. Seki, A. Shimada, Y. Hayashi, T. Morio, S. Kumaki, Y. Ishida, Y. Kamachi, and A. Yachie. 2006. Omenn syndrome--review of several phenotypes of Omenn syndrome and RAG1/RAG2 mutations in Japan. Allergol. Int. 55: 115-119.
- Jones, J. M. and C. Simkus. 2009. The roles of the RAG1 and RAG2 "non-core" regions in V (D) J recombination and lymphocyte development. Arch. Immunol. Ther. Exp. (Warsz) 57: 105-116.
- Notarangelo, L. D., M. S. Kim, J. E. Walter, and Y. N. Lee. 2016. Human RAG mutations: biochemistry and clinical implications. Nat. Rev. Immunol. 16: 234-246.