


|
|
DOI Prefix 10.20431 |
Information
Journal Policies
Secondary Mitochondrial Genome Deletions in Hereditary Spastic Paraplegia due to SPG7 Mutations
Caroline Passaplan, BM1, Ramona Bolognini2, Sabina Gallati2, Jean-Marc Burgunder, MD1,3* Andre Schaller, MD2,4*
2.Department of Medical Genetics, University of Bern, Switzerland.
3.Neurozentrum Siloah, CH-3073 Gümligen, Switzerland.
4.Medizinische Genetik, Kinderspital, Inselspital, CH-3010 Bern, Switzerland.
Copyright : © 2018 . This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Hereditary spastic paraplegia is a heterogeneous group of diseases due to mutations in more than 50 different genes. One of the most common autosomal recessive forms is found in mutations of the paraplegin gene, and presents as a complex phenotype. However, clinical aspects of molecular mechanisms involved in paraplegin mutations remain to be explored.
We have examined a family with a phenotype suggestive of a mitochondrial cytopathy by clinical methods including muscle biopsy and genetic evaluation in muscle and circulating leukocytes.
An autosomal recessive presentation was found with a slowly progressive disorder including myopathy, chronic progressive external ophthalmoplegia, spastic paraplegia, and cognitive impairment. Mitochondrial DNA deletions of different sizes were found at high levels in muscle biopsies. Genotyping disclosed a homozygous deletion (c.1454_1462) mutation in the paraplegin gene (SPG7) in exon 11.
These data demonstrate a profound overlap between the phenotype of SPG7 and mitochondrial cytopathy. This is explained by secondary mitochondrial deletions due to paraplegin gene mutations. The molecular mechanism of this effect needs to be further explored.
Paraplegin, SPG7, Mitochondrial gene deletion, myopathy, spastic paraplegia, ptosis, executive disorder, depression.
HSP: hereditary spastic paraplegia
PCR: polymerase chain reaction
SPG7: Spastic paraplegia 7 gene
SPRS: spastic paraplegia rating scale
SARA: scale for the assessment and rating of ataxia
1. Introduction
Hereditary spastic paraplegia (HSP) is a heterogeneous group of diseases with varied clinical symptoms, age of onset and progression. HSP may present with diverse forms of inheritance and mutations in more than 70 genes are known to cause various forms of the disorder[1]. HSP is divided into pure and complicated forms. Spasticity and a weakness in lower limbs, a diminished vibratory sense in the lower limbs and a urinary urge characterize the pure form of the disease. The complicated form presents as a complex phenotype with involvement also in upper limbs, peripheral neuropathy, amyotrophy, cognitive impairment, cerebellar dysfunctions, ocular troubles and extrapyramidal features[2, 3].
The presentation of a complex form in HSP has some similarities with some mitochondrial cytopathies, which typically may present with a myopathic involvement including external eye and skeletal muscles accompanied by encephalopathic and spinal involvement leading to cognitive disorders and spastic paraplegia[4].
Here, we report on a family with chronic progredient external ophthalmoplegia accompanied by skeletal myopathy, spastic paraplegia, cognitive changes due to a homozygous mutation in the paraplegin gene SPG7 associated with secondary mitochondrial deletions.
2. Material And Methods
2.1. Patients
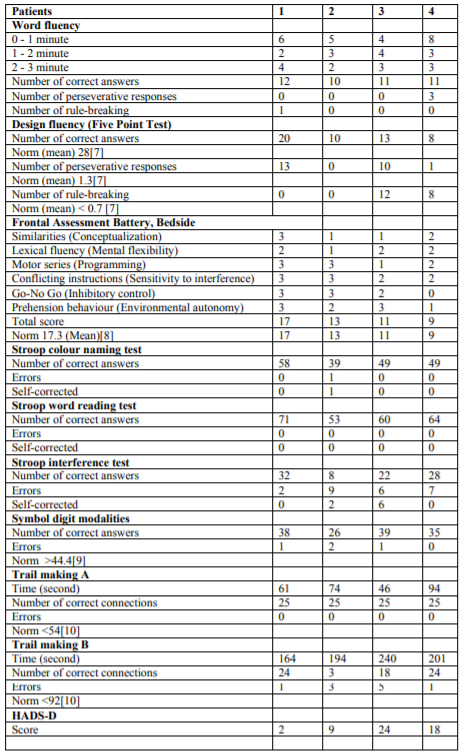
A family from Western Switzerland was examined, clinical investigations included a battery of standard neurological assessment, including the spastic paraplegia rating scale (SPRS)[5]. In order to be able to assess additional signs not captured by the SPRS, and also to assess potential impairment for the performance of the neuropsychological tests mentioned below, the scale for the assessment and rating of ataxia (SARA)[6] was also included. Furthermore, in the assumption, that prefrontal involvement might be included in addition to the frontal lesion leading to spastic paraplegia, a battery of neuropsychological tests including assessment of executive function has been included. Following neuropsychological tests were performed: phonemic fluency test (one letter for 3 minutes), five point design fluency test[7], frontal assessment battery at bedside[8], Stroop test (colour naming, word reading, and interference test), symbol digit modalities test[9], and trail making test[10]. Depression and anxiety were assessed by the Hospital Anxiety and Depression scale. Additional investigations have selectively included laboratory testing, and magnetic resonance imaging of brain and spine. Muscle biopsy was performed in some cases and examined by light microscopy with HE and GT staining, oil-red-oil and enzyme histochemistry (NADH, ATP at pH 4.3, 4.6, 9.0, esterase, COX). Muscle histology was qualitatively assessed visually. The study was approved by the State Ethics Board (Kantonale Ethik Kommission, Bern), and all patients have provided informed consent.
Total DNA extraction from EDTA-blood and skeletal muscle was performed according to standard isolation protocols (Qiagen). Large- scale rearrangements were analysed by long polymerase chain reaction (PCR) as described previously[11]. mtDNA was screened for mutations by PCR and subsequent single strand conformation polymorphism as previously described[12, 13]. The screening of nuclear genes involved in mitochondrial deletions included the coding parts (exons), including the exon-intron boundaries of POLG1 (23 exons), POLG2 (8 exons), ANT1 (4 exons) and Twinkle (5 exons).
For MitoExome sequencing of the index patient an in-solution hybridization capture method (NimbleGen) was used which was subsequently sequenced on a illumina HiSeq 2500. The 4.2 Mb of targeted DNA included all coding and untranslated regions of 1476 nuclear genes including 1013 mitochondrial genes from the MitoCarta database[14] and 463 additional genes. The sequencing was performed with a paired-end approach. Reference alignment was done with CLC Workbench v.7.0.3. Mapped reads were subsequently used for variant calling.
3. Results
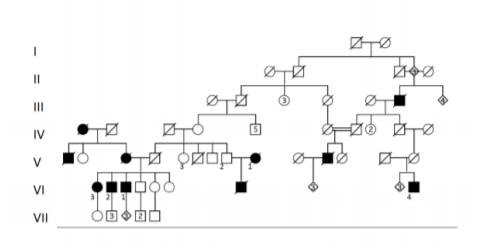
The index case was a 62-years old former agricultural engineer (Figure 1: VI.2), who had had eyelid ptosis for many years. The first symptoms started at 54 years of age with muscle pain, intolerance at work efforts and muscle weakness. He gradually developed chronic back pain, muscle cramps and migraines. His gate worsened and spasticity was manifest about 3 years later with impaired balance and falls gradually developed. His relatives mentioned changes in the eye movements at that time. Later he experienced tinnitus and dysarthria related to cold temperature. He had also several episodes of depression. The gradual course of progression led to increased functional impairment with change in work abilities.
At examination he had a muscular weakness of the face with severe bilateral ptosis and external ophthalmoplegia. Fundoscopy, visual acuity and pupillary reflex was normal. Diadochokinesia was disturbed and the finger-to-nose test was dysmetric, the heel to knee test was not possible. The muscle tone was spastic with brisk reflexes and some rigidity, much more severe in the legs and a positive Babinski sign. There was a proximal weakness with muscle atrophy, more in the legs. The sensory examination was normal except for a diminished vibratory sense in the legs. Gait was severely impaired due to spasticity and slight ataxia. Cognitive functions where slightly decreased.
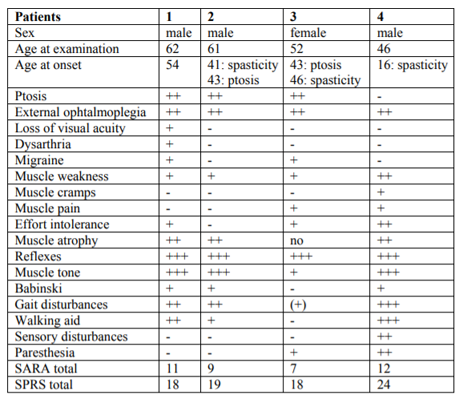
A number of other cases of gait impairment and/or ptosis were reported in the family in 4 generations, with instances of consanguinity (Figure 1). Several members of the family could be examined, most of them had spasticity, myopathic features and external ophthalmoplegia. The symptoms had started a similar ages in the brother and sister of the index case. However a further relative had an earlier onset of spasticity with 16 years old, with muscle cramps and progression over a few years to unability to walk. The most common features include external ophthalmoplegia with or without ptosis, prominent lower limbs spasticity, sensibility disorders, stiffness in the lower limbs and progressive gait disturbance accompanied by myopathic impairments like muscle pain and muscle atrophy and loss of strength (Table 1). Other members of the family, who could not be examined, were reported to have had various degrees of ptosis (in some instances seen on old photos) and gait disorder, including weakness and stiffness, which could not be well differentiated from history. There is consanguinity in the family with more persons from the same small geographical area married into the family.
Differences in symptoms between the three examined siblings (VI.1; VI.2 and VI.3) are related to age, being more severe in the older brother. The patient of another branch of the family, shows another pattern of age and symptoms at onset: he developed weakness and progressive gait disturbance in the both legs much earlier and spastic paraplegia is more severe. Furthermore, this patient has problem with fecal incontinence since 2-3 years, what was not found in the others. External ophthalmoplegia was seen but without eyelid ptosis. VI.1; VI.2 and VI.3. The total score of the SARA (Table 1) was in the low abnormal range for all patients examined, due to high abnormal values in gait and stance items. The total score of the SPRS (Table 1) was in the middle abnormal range for all patients examined. All patients showed cognitive impairment in low to medium range (Table 2). The word fluency was impaired in similar was in all patients. The two patients had a severe and one a mild state of anxiety and depression (Table 2).
All 4 examined affected cases had tried different drug treatment. Baclofen had been of help in one against pain. All mentioned that spasticity and leg pain are improved by stretching movements and regularly muscles exercises. No myopathic change were found at light microscopy in any of the three muscle biopsies performed. ATP-ase histochemistry showed a normal checkboard pattern of coloration outside of discrete chronic denervation signs found in the muscle biopsy of patient one. No changes in oxidative enzymes were found by using NADH-TR and cytochrome oxidase staining. There was no fat accumulation in the oil-red-oil staining.
No single point mutations of the mitochondrial genome were found in muscle tissues of the three examined biopsies. However, multiple deletions were found in all of them. The size of the deletion was 3-12 kpb with a hetereroplasmy of 98% in VI.2 case, 2-10 kbp with a heteroplasmy of 35-70% in the VI.1, and 6-10kpb with a 54% of heteroplasmy in the VI.4. No mutation was found in POLG1, POLG2, ANT1 and Twinkle.
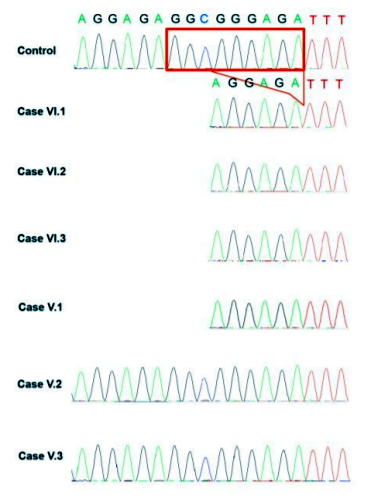
A homozygous deletion within the SPG7 gene was found, this leads to an in frame deletionc.1454_1462delGGCGGGAGA (p.Glu484_Arg486del) of the (Exon 11) (Figure 2).
Using Sanger sequencing the mutation was confirmed in the index patient and in all affected family members (Figure 1: VI.1; VI.2; VI.3; VI.4).
4. Discussion
The clinical features of this family with external ophthalmoplegia, myopathic symptoms including weakness, atrophy, exercise intolerance, cramps, along with spasticity, migraine and cognitive impairment are quite suggestive of a mitochondrial disorder. The involvement of the mitochondrial genome was demonstrated by the presence of multiple deletions in all the muscle biopsies studied. According to a practical molecular-genetic classification of the mitochondrial disorders[4], these findings place these observation among the category of defects in mitochondrial DNA (mtDNA) maintenance. Other categories include the mitochondrial gene mutations and mutations in nuclear genes coding for mitochondrial proteins. The presence of inherited external ophthalmoplegia by itself already strongly suggests a disorder of mtDNA maintenance. External ophthalmoplegia is often accompanied by other clinical features and is frequently due to mutations in nuclear genes involved in mtDNA replication and repair, including POLG, Ant, and Twinkle.
Next generation sequencing has led to the recognition an increasing number of additional genes involved in complex mitochondrial disorders with impaired mtDNA maintenance. Using such a strategy, we found a mutation in the SPG7 gene encoding paraplegin. The homozygous presence of this mutation is compatible with the consanguinity found in the family. The mutation is found in a gene region of a known disease associated mitochondrial gene and is not listed in dbSNP137[15]. This mutation has already been reported[16, 17] and is one of the most frequent SPG7 mutations[18]. The phenotype is quite variable, with a relatively pure upper motoneurone type of presentation[17] or a more complex presentation including ataxia[18]. A prominent presentation in our series is the involvement of external eye muscles with external ophtalmoplegia and ptosis. Subtle eye movements have been described in earlier series[18], but since they are rare, it has been suggested that they represent a coincidental finding, our data demonstrate, that this may not be the case.
In our cases, muscle histology did not demonstrate confirmatory findings of a mitochondrial disorder in contrast this to some previous studies, which had shown cytochrome oxydase (COX)-deficient fibres and ragged-red fibres[16, 18, 19]. This lack of correlation between the intensity of histological changes and the presence of mtDNA deletion is not quite clear but points at the importance of a direct mtDNA molecular genetic analysis using appropriate techniques. An earlier study had searched for mtDNA changes but did not find any by Southern blot[20]. Our finding of multiple deletions with variable numbers is in line with those of two recent studies[21, 22]. New SPG7 mutations were recently found in a group of patients with progressive external ophthalmoplegia. Their muscle biopsies included some COX deficient fibres and ragged red fibres[22]. Our cases presented with a quite similar phenotype as the one described in that study. They also had a mid-adult life onset of progressive external ophtalmoplegia, ptosis, and spastic paraparesis, however, they did not have dysphagia, only one of them suffers of dysarthria, and we did not find optic atrophy or bladder problems. Similar findings were made in two Norvegian families with chronic external ophtalmoplegia, ptosis and hereditary spastic paraplegia with a homozygous, respectively a compound heterozygote SPG7 mutation[21]. In the family presented here, the affected patients have cognitive impairment. The psychological and cognitive tests we made show disorders of memory, concentration, processing speed and executive functions. The patients have furthermore depressive symptoms. These data confirm the complexity of the phenotype, however, the variety is not yet explained by the genetic data available and modifying genetic or environmental factors may play a role.
About 1,5-6 % of cases with hereditary spastic paraplegia are caused by a mutation of the paraplegin gene[21] mapped on the chromosome 16q24.3 and more than 50 pathogenic mutations of this gene have been described. Paraplegin is a subunit of an ATP-dependant m-ATPase located in the inner membrane of the mitochondria, leading to the suggestion, that mutations of this gene would directly impair mitochondrial function and explain the histological findings are responsible for mitochondrial disorders; in some cases of patients with SPG7 mutations, muscle biopsies showed signs of mitochondrial disorders with ragged-red fibres and cytochrome c negative fibres in the muscular tissues[23].
These data demonstrate that SPG7 mutations may cause accumulation of mitochondrial DNA damages and multiple respiratory chain changes in muscles. This includes paraplegin mutations to the list of the most common genes involved in mtDNA maintenance disorders and in mitochondrial replication[24] with Twinkle[25, 26], ANT1[26], POLG1[27], POLG2, OPA1[28], TYMP[29], RRM2B[30]. This has some implications for the guidance of molecular-genetic testing in patients with these complex phenotypes.
5. Declarations
Ethics Approval and Consent to Participate: The Ethics Board of the Canton of Bern, has approved the study. Patients have provided informed consent for participation in the study.
Consent for Publication: All authors have provided consent fort the publication of the manuscript.
Availability of Data and Material : Data not allowing patient identification are in the main portion of the manuscript
6. Acknowledgements
The authors are thankful to the patients for their participation in the study.
5.References
- Fink JK. Hereditary spastic paraplegia: clinical principles and genetic advances. Semin Neurol. 2014 34: 293-305.
- Finsterer J, Loscher W, Quasthoff S, Wanschitz J, Auer-Grumbach M, Stevanin G. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci. 2012 318: 1-18.
- Lo Giudice T, Lombardi F, Santorelli FM, Kawarai T, Orlacchio A. Hereditary spastic paraplegia: clinical-genetic characteristics and evolving molecular mechanisms. Exp Neurol. 2014 261: 518-539.
- Dimauro S, Schon EA, Carelli V, Hirano M. The clinical maze of mitochondrial neurology. Nat Rev Neurol. 2013 9: 429-444.
- Schule R, Holland-Letz T, Klimpe S, et al. The Spastic Paraplegia Rating Scale (SPRS): a reliable and valid measure of disease severity. Neurology. 2006 67: 430-434.
- Schmitz-Hubsch T, du Montcel ST, Baliko L, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology. 2006 66: 1717-1720.
- Tucha L, Aschenbrenner S, Koerts J, Lange KW. The five-point test: reliability, validity and normative data for children and adults. PLoS One. 2012 7: e46080.
- Dubois B, Slachevsky A, Litvan I, Pillon B. The FAB: a Frontal Assessment Battery at bedside. Neurology. 2000 55: 1621-1626.
- Sheridan LK, Fitzgerald HE, Adams KM, et al. Normative Symbol Digit Modalities Test performance in a community-based sample. Arch Clin Neuropsychol. 2006 21: 23-28.
- Tombaugh TN. Trail Making Test A and B: normative data stratified by age and education. Arch Clin Neuropsychol. 2004 19: 203-214.
- Kleinle S, Wiesmann U, Superti-Furga A, et al. Detection and characterization of mitochondrial DNA rearrangements in Pearson and Kearns-Sayre syndromes by long PCR. . Hum Genet. 1997 100: 643-650.
- Kleinle S, Schneider V, Moosmann P, Brandner S, Krähenbühl S, Liechti-Gallati S. A novel mitochondrial tRNA(Phe) mutation inhibiting anticodon stem formation associated with a muscle disease. Biochem Biophys Res Commun. 1998 9: 112-115.
- Liechti-Gallati S, Schneider V, Neeser D, Kraemer R. Two buffer PAGE system-based SSCP/HD analysis: a general protocol for rapid and sensitive mutation screening in cystic fibrosis and any other human genetic disease. Eur J Hum Genet. 1999 7: 590-598.
- Pagliarini DJ, Calvo SE, Chang B, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008 134: 112-123.
- Lieber DS, Calvo SE, Shanahan K, et al. Targeted exome sequencing of suspected mitochondrial disorders. Neurology. 2013 80: 1762-1770.
- McDermott CJ, Dayaratne RK, Tomkins J, et al. Paraplegin gene analysis in hereditary spastic paraparesis (HSP) pedigrees in northeast England. Neurology. 2001 56: 467-471.
- Brugman F, Scheffer H, Wokke JH, et al. Paraplegin mutations in sporadic adult-onset upper motor neuron syndromes. Neurology. 2008 71: 1500-1505.
- van Gassen KL, van der Heijden CD, de Bot ST, et al. Genotype-phenotype correlations in spastic paraplegia type 7: a study in a large Dutch cohort. Brain. 2012 135: 2994-3004.
- Casari G, De FM, Ciarmatori S, et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998 93:973-983.
- Arnoldi A, Tonelli A, Crippa F, et al. A clinical, genetic, and biochemical characterization of SPG7 mutations in a large cohort of patients with hereditary spastic paraplegia. Hum Mutat. 2008 29: 522-531.
- Wedding IM, Koht J, Tran GT, et al. Spastic paraplegia type 7 is associated with multiple mitochondrial DNA deletions. PLoS One. 2014 9: e86340.
- Pfeffer G, Gorman GS, Griffin H, et al. Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain. 2014 137: 1323-1336.
- Klebe S, Depienne C, Gerber S, et al. Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy. Brain. 2012 135: 2980-2993.
- Copeland WC. Defects in mitochondrial DNA replication and human disease. Crit Rev Biochem Mol Biol. 2012 47: 64-74.
- Spelbrink JN, Li FY, Tiranti V, et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet. 2001 28: 223-231.
- Kaukonen J, Juselius JK, Tiranti V, et al. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science. 2000 289: 782-785.
- Van Goethem G, Dermaut A, Löfgren J-j, Van Broeckhoven M, Christine. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nature Genetics. 2001 28: 211-212.
- Jonasdottir A, Eiberg H, Kjer B, Kjer P, Rosenberg T. Refinement of the dominant optic atrophy locus (OPA1) to a 1.4-cM interval on chromosome 3q28-3q29, within a 3-Mb YAC contig. Hum Genet. 1997 99: 115-120.
- Hirano M, Lagier-Tourenne C, Valentino ML,Marti R, Nishigaki Y. Thymidine phosphorylase mutations cause instability of mitochondrial DNA. Gene. 2005 354: 152-156.
- Bourdon A, Minai L, Serre V, et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007 39: 776-780.