


|
|
DOI Prefix 10.20431 |
Information
Journal Policies
ARC Journal of Neuroscience
Volume-1 Issue-1, 2016
Abstract
The alternative splicing (AS) mechanism is considered the major driving force of transcriptome and proteome diversity. Itrelies on a delicate and finely tuned interplay among a great number of molecular elements. The crucial action of AS in the regulation of diverse biological processes is not limited to physiological states, but is mirrored in the growing list of human diseases associated with known or suspected splicing defects, including neurodegenerative and oncologicaldiseases. In these pathologies, the AS regulation of PARK2gene (also called parkin RBR E3 ubiquitin protein ligase), one of the largest in our genome, seems to play a fundamental role. Here, we will briefly review some major data concerning the genetic organization, the transcription regulation, the structure of the protein and the relative molecular functions of PARK2. Then we will focuson the current knowledge about PARK2 alternative spliced isoforms and their implication in human diseases.
2.KEYWORDS
3. INTRODUCTION
4.PARKIN CYTOGENETIC LOCATION, GENE STRUCTURE, SPLICING ISOFORMS AND PROTEIN FUNCTIONS
5.PARKIN ALTERNATIVE SPLICING IN PHYSIOLOGICAL CONDITIONS
6.PARKIN ALTERNATIVE SPLICING: INVOLVEMENT IN NEUROLOGICAL DISORDERS
7.PARKIN ALTERNATIVE SPLICING: INVOLVEMENT IN CANCER
8.CONCLUSIONS
9.ACKNOWLEDGEMENTS
10.REFERENCES
AUTHOR DETAILS
Lajya Devi Goyal
KEYWORDS
PARK2, parkin, alternative splicing, Parkinson’s disease, cancer.
INTRODUCTION
Aalternative splicing (AS) is a fundamental mechanism of gene expression regulation that involves more than 90% of genes in our genome [1, 2]. This process is considered the major driving force of transcriptome and proteome diversity: thanks to its ability in increasing the coding potential of genomes, AS represents a cheap and powerful tool that allows cells to produce multiple protein products starting from a single gene. Through AS, the immature primary mRNA transcript can be processed and combined in different ways by alternative usage of exons, splice sites or intron sequences[3].
The AS mechanism relies on a delicate and finely tuned interplay amonga great number of molecular elements: the constitutive splicing motifs (i.e. the 5’ and the 3’ splice sites, the lariat branch point and the polypyrimidine tract), the splicing regulatory sequences (i.e. exonic and intronic splicing enhancers or silencers), the components of the spliceosome (i.e. the small nuclear ribonucleoproteins U1, U2, U4, U5, and U6) and other additional auxiliary RNA-binding proteins[4].
Besides the physiological involvement of AS in cellular life, many evidences suggest a significant contribution of this mechanism in human pathologies[5] and therefore its clinical relevance is growing exponentially. It is estimated that 50% of disease-causing mutations affect pre-mRNA splicing[6]. Two main molecular defects can lead to aberrant AS events: mutations of the DNA elements required for correct pre-mRNA processing (called cis-acting mutations) oralterations of the factors that are necessary for splicing regulation (termed trans-acting defects).
ThePARK2(or parkin RBR E3 ubiquit in protein ligase)gene belongs to the family of extremely large genes and is located in the long arm of chromosome 6 (6q25.2-q27). Mutations of this gene were firstly described as responsible for familial forms of Parkinsonism, but they also occur in a wide variety of human diseases, including pathologies of the nervous system and malignancies. Our knowledge about PARK2AS regulation is enormously increased from the cloning of the first human cDNA [7 8].A remarkable number of papers currently demonstrated, in human as well as in other species, the existence of different mRNA parkin variants [9- 15], potentially encoding a wide varieties of isoforms with different structures and molecular architectures. Thanks to this renewed update regarding PARK2 AS, several papers are addressing the experimental study with the awareness of this complex splicing mechanism [16-22].
In the present review, we will briefly describe some major data on PARK2, concerning the genetic organization, the transcription regulation, the structure of the parkin protein and relative molecular functions. Then we will summarize the current knowledge about PARK2 alternative spliced mRNAs and protein isoforms and their implication in human diseases.
PARKIN CYTOGENETIC LOCATION, GENE STRUCTURE, SPLICING ISOFORMS AND PROTEIN FUNCTIONS
PARK2 is one of the largest gene in human genome and encompasses more than 1.38 Mb of genomic DNA in the long arm of chromosome 6 [7, 8] (Figure 1). It is located in the center of the third most mutation-susceptible common fragile site (called FRA6E), a genomic region characterized by intrinsically difficulties to replicate and prone to forming chromosomal breakages [23]. Although FRA6E contains many genes, the main fragility coreis localized within the PARK2 gene sequence. Inside the FRA6E locus, PARK2 is flanked towards the telomeric direction by PACRG (or PARK2 co-regulated), which lies in a head-to-head arrangement and shares a common promoter with the adjacent PARK2 [24], and in the centromeric direction by AGPAT4 (1-acylglycerol-3-phosphate O-acyltransferase), encoding a member of the 1-acylglycerol-3-phosphate O-acyltransferase family. According to NCBI Map Viewer, further elements overlap or surround the PARK2 genetic region, such as two pseudogenes (KRT8P44 and TRE-TTC15-1) and a set of non-coding RNAs (LOC105378094, LOC105378098, LOC105378097 and LOC105369171)(Figure 1).
Figure1. Cytogenetic location of PARK2 gene and surrounding region.
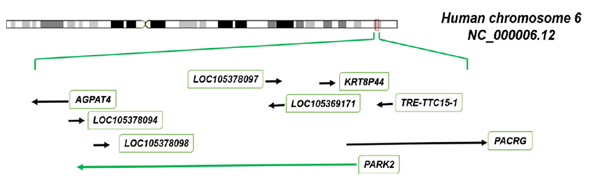
The figuredisplays the human chromosome 6 (NCBI Reference Sequence NC_000006.12) with relative localization and flanking genes of PARK2. The orientation of PARK2 on the chromosome is represented by agreen arrow and transcribed in antisense direction (reverse strand). Itis flanked towards the telomeric direction by PACRG in a head-to-head arrangement and in the centromeric direction by AGPAT4. Further elements overlap or surround the PARK2 genetic region, such as two pseudogenes (KRT8P44 and TRE-TTC15- 1) and a set of non-coding RNAs (LOC105378094, LOC105378098, LOC105378097 and LOC105369171).
The first human PARK2 transcript was isolated in 1998 by Kitada et al. [7]. It consists of a 2,960 nucleotides transcript with a 1,395-base-pair open reading frame encoding a protein of 465 amino acids [7]. Based on this mRNA, the genomic organization and exon/intron boundary sequences of PARK2was established of 12 exons [7]. From that first cloned cDNA, PARK2 gene has been the focus of hundreds of scientific papers which concentrated both on the genetic variations present in the 12 originally established exons and in their exon/intron boundaries. However, a number of transcripts and additional exonic sequences alternatively included or skipped in mature mRNAs have been collected during years.
The main works of recapitulation of PARK2 human transcripts and protein isoforms have been performed by our own research group [14, 19,20]. In the first one, published in 2004, seven PARK2 splice transcripts were isolated from brain or human leukocytes and were indicated as TV1, TV2, TV3, TV6, TV7, TV11 and TV12(Table 1). More recently, we have reviewed data [19], showing that the Homo sapiensPARK2 cDNA sequences, deposited in Uni Gene repository, aligned with the genomic sequence and clustered in a minimal non-redundant way, support at least the existence of 21 different alternatively spliced mRNAs (named from H1 to H21) composed of 17 exons[19, 20](Figure 2 and Table 1).For each of these 21 variants, the protein amino acidic sequence (based on the longest reading frame), molecular weight, isoelectric point and domain composition were predicted[19, 20]. Despite the multiple described splicing variants, the NCBI Reference Sequence database currently annotates just three representative transcripts as full-length PARK2 mRNAs (NM_004562; NM_013987; NM_013988) encoding proteins of 465, 437 and 316 amino acids respectively (Table 1).
The table reports the NCBI Accession Numbers, the Gene Identifiers (GI) and the mRNAs code name assigned in [14] and[19]. The corresponding amino acidic length and predicted molecular weight are also indicated.
Figure2. Exonic and intronic organization of human PARK2 gene.
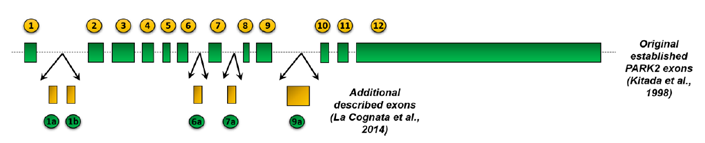
The figure shows the genetic organization of human PARK2: the 12 originally identified exons[7] are represented as green bars. 5 additional exonic regions have been described by La Cognata et al., 2014 [19] and are colored in yellow. These 5 additional exons have been renamedas exons 2, 3, 9, 11 and 14. The size of all exons and introns (black line) is proportional to their length.
The full-length parkin is widely expressed in different brain areas[25] and participates in the ubiquitin proteasome system as a RING-type E3 ubiquitin-ligase. This participation is accomplished at its N terminus through a ubiquitin-like domain (UBL) recognizing a specific substrate protein and four zinc-coordinating RING-like domains: RING0,RING1, IBR and RING2[26] (Figure 3). The UBL domain is involved in recognition and transfer of ubiquitin to specific substrate proteins, thereby targets them for proteasome degradation[26]. Various types of proteins, including cytosolic (Synphilin-1, Pael-R, CDCrel-1 and 2a, α-synuclein, p22, Synaptogamina XI) [27-31], nuclear (Cyclin E) [9], and mitochondrial ones (MFN1 and MFN2, VDAC, TOM70, TOM40 and TOM20, BAK, MIRO1 and MIRO2, FIS1) [32-36] have been identified as parkin substrates.The number of targets is such high that parkin protein results involved in numerous molecular mechanisms(proteasome- degradation, mitochondrial homeostasis, mitophagy, mitochondria
Figure3. Structural domain composition of the full-length Parkin protein
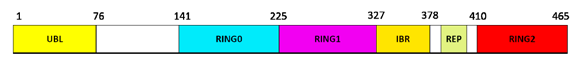
Parkin consists of an ubiquitin-like domain (Ubl) at its N terminus and four zinc-coordinating RING-like domains: RING0, RING1, IBR and RING2. The REP domain represents the repressor element of parkin (REP), so-called for its role in the regulation of parkin activity [26]. Numbers indicates the relative start and stop amino acidic positions.
PARKIN ALTERNATIVE SPLICING IN PHYSIOLOGICAL CONDITIONS
ThePARK2regulation represents a fascinating example of the use of AS to create different variants within single cell types. Indeed, the PARK2 AS intervenes in normal physiological condition during cell life. Some evidences demonstrating common PARK2 variants could be cited: in the first original paper, Kitada et al. [7] described a cDNA clone without a small sequence of 84 bp in frame revealed by screening human fetal brain and skeletal muscle cDNA libraries; our own group (as anticipated) reported seven PARK2 splice transcripts isolated from brain or human leukocytes[14]; Sunada et al. demonstrated a parkin exons 3-4-5 (447 bp; 149 AA) splice variant (called E3-4-5SV) expressed in human leukocytes[15]; Kasap et al. [37], showedthat the protein is expressed in serum and furthermore, identified six different proteins with similar molecular weight, probably corresponding to spliced forms, through MALDI-TOF analysis of the immunore active area cutted off from the blot.
The interest onPARK2 AS becomes obviously greater when it concerns human diseases: the differential expression of PARK2splicing isoforms have been studied in different pathological states, as we will discuss in the next paragraphs.
PARKIN ALTERNATIVE SPLICING: INVOLVEMENT IN NEUROLOGICAL DISORDERS
Homozygous or compound heterozygous mutations in PARK2 are the most common cause (50% of cases) of autosomal recessive juvenile Parkinsonism (AR-JP), a form of early-onset characterized by classic symptoms of PD (such as bradykinesia, rigidity, and tremor), a clear drop in onset age, a slow disease progression and a good response to levodopa treatment [38]. PARK2 mutations also explain ~15% of the sporadic cases with onset before 45 [39, 40] and act as susceptibility alleles for late-onset forms of PD (2% of cases) [41]. Along with the about 200 mutations currently identified in PARK2 coding region, a number of cis-acting point mutations in splice acceptor or donor sites have been observed in PD patients [19, 42 -48].These are collected in the Parkinson Disease Mutation database (http://www.molgen.vib-ua.be/PDMutDB/).
Several evidences support the differential expression of PARK2 splice variants in PD conditions. A parkin splice transcript, called by authors E4SV and lacking exon 4 (122 bp) [13], was identified in the substantia nigra and leukocytes of sporadic PD patients and healthy controls by reverse transcriptase-polymerase chain reaction. The loss of exon 4resulted in a reading frame shift over the junction of exons 3-5 and a stop codon 17 bp downstream from exon 3, producing a truncated protein with a total loss of the two-RING finger functional domain. Although both groups expressed these variants, the authors highlighted a significant over-expression of E4SV and reduction of the wild-type parkin in the leukocytes of sporadic PD patients as compared to age-, gender-, and race-matched controls[13]. Another study conducted by Humbert, Beyer and colleagues [11, 12], was aimed to explore the differential expression of PARK2 splice variants in different neurodegenerative diseases, including PD. Of seven analyzed parkin isoforms, only two splice variants (Parkin TV3 and TV12) showed significant over expression levels in PD frontal cortex when compared to controls.
Lewy body diseases comprise a group of disorders characterized by the presence of neuronal inclusions called Lewy bodies. This group comprehends PD and two different forms of dementia with LBs (DLB): pure DLB (pDLB) and common Lewy body disease (cLBD),(i.e. a mixed form of pDLB with the widespread presence of LBs and Alzheimer disease because of the additional presence of amyloid plaques).
The differential expression of seven parkin isoforms were assessed in frontal cortex of patients with pLBD and cLBD. At least four transcripts showed altered levels compared to controls. Parkin isoform TV1, the unique whole transcript of the gene with preserved complete function as E3 ubiquitin-ligase, showed altered expression levels and was down regulated in both pDLB and cLDB, suggesting a substrate accumulation of material by insufficient ubiquitylation. In addition, Parkin TV7 relative expression levels revealed a marked decrease in both Cdlb and DLB when compared to controls. Anover expression of bothTV2 and TV3 were only seen in cDLB. Both of them present a significant shortening of the sequence betweenthe ubiquitin-like domain and the ring-finger domains that could alter substrate affinity and/or recognition, resulting in insufficient degradation of parkin substrates and subsequent LB-like inclusion formation.
The expression of parkin splice variants was explored in the frontal cortex of AD patients. Despite the observed different quantitative measurements among the spliced parkin variants, no significant disease-specific isoform-expression changes were revealed[11, 12]. Parkin splicing, therefore, seems to be not involved in the pathogenesis of AD.
Together with PD and DLB, multiple system atrophy (MSA) is a member of the neurodegenerative disorders termed α-synucleinopathies. In a very recent study[49], the differential expression of parkin transcript isoforms in different brain areas (substantia nigra, striatum, cerebellar cortex, nucleus dentatus and prefrontal cortex) was ascertained in MSA patients in comparison to PD cases and healthy controls. Authors reported in MSA subjects increased levels of parkin isoforms lacking the N-terminal ubiquitin-like domain. In particular, the transcription of the TV1 variantwas reduced in MSA substantia nigra and PD prefrontal cortexas compared to normal controls, suggesting a loss of the parkin ability to promote degradation, ubiquitination and relative neuroprotective activity. Moreover, higher levels of TV2 transcripts were found in the striatum and cerebellar cortex of MSA patients as compared to PD and normal subjects. There were also higher levels of the TV12 splice variant mRNA in the substantia nigra, nucleus dentatus, striatumand cerebellar cortex of MSA as compared to PD and control brains. The changed expression profile of these isoforms suggested regional and cellular alterations in the splicing events that may be important for the highly increased aggregation of α-synuclein in the brain.
PARKIN ALTERNATIVE SPLICING: INVOLVEMENT IN CANCER
Parkin gene has been shown to be genetically altered in a wide variety of human tumors including lung cancer. Its germline and somatic deletions were reported both in patients and in lung adenocarcinoma cell linessuggesting that the loss of this locus and the resulting changes in the expression are involved in the development of this tumour [50] [51]. Very recently, Xiong et al. reported the PARK2 germline mutation c.823C>T (p.Arg275Trp) in a family with an heredited form of lung cancer [52].
The expression pattern of parkin spliced isoforms has been investigated in deparaffinized sections of human lung adenocarcinomas sampleswith western blot analysisby using two different antibodies recognizing distinct protein domains[21]. In tumoural samples, bands of ~50, ~42 and ~20kDa molecular weight were detected on blot, corresponding to H1/H5, H14/H4/H8/H17 and H13/12 isoforms, respectively. Moreover, in order to characterize the biological role of these proteins, the authors have assessed their expression in human lung adenocarcinoma (A549) and in human normal bronchial epithelial cell lines (BEAS-2B) after treatment with specific stressors. They showed that parkin isoforms expression profile changed after exposure to a proteasome inhibitor or mitochondrial depolarizing agent, or in serum starvation. Results suggested that some of them may be involved in specific cellular processes, such as mytophagy or apoptosis.
PARK2 has been decribed as a tumour suppressor gene in glioblastoma multiforme, the most commonand lethal primary malignancy of the central nervous system[22, 53]. Genetically, both copy number loss and several somatic point mutations of parkin have been describedin glioblastomas samples[53]. Transcriptionally, itsexpression was found dramatically reduced in human glioma cells, while its restoration promoted cell-cycle arrest and mitigated their proliferation rate [54] [55].
In order to evaluate the involvement of parkin proteins in this tumor malignancy,theirexpression profile was investigated in different grades astrocytomas [22]. In all tumor samples were detected bands of ~58, ~50 and ~15 kDa kDa molecular weight on blot, corresponding to H20, H1/H5 and H9/H13/H7/ H18 isoforms, respectively. Furthermore, in afrozen sample of glioblastoma multi forme, a further band of ~42 kDa molecular weight corresponding to H14/H4 isoforms, was also visualized by western blot analysis. Densitometric analysis of blot showed that parkin isoforms expression was higher in malignant glioblastomas than in less invasive gliomas. Furthermore, to investigate the role of splice isoforms in cellular processes, it has been analyzed their expression in three glioblastoma cell lines (T98G, A172 and U87-MG) following treatment with a proteasome inhibitor, or induction of mitophagy, or after serum deprivation. Results suggest that expression of the isoforms changed after treatments in cell specific manner.
The regulation of Parkin splice variants expression was investigated also in clinical specimens of human colorectal cancer tissues[9]. The authors identified various alternatively spliced isoforms of Parkin in human colorectal cancers, mainly constituted by loss of exons 3-6, and functionally defective in their ability to induce proteolytic degradation of the cyclin E protein. These findings suggest that the AS of the Parkin gene might contribute to the accumulation of cyclin E protein, leading to the deregulation of cell- cycle progression in colorectal cancers. In support of these results, the expression of alternatively spliced Parkin was also measured in colon cancer cells: DLD1 cells expressed a Parkin transcript lacking exons 3 to 5 and exons 7-8, while HT29 colon cancer cells expressed predominantly wild-type Parkin.
In order to clone the FRA6E site and better characterize the enclosed genes, Denison et al.[56] performed a screening of PARK2 splicing variants in normal ovarian epithelium, primary ovarian tumors and ovarian cancer cell lines. This screening revealed several alternative transcripts in both the cell lines and the primary tumors analyzed, but not in the healthy ovarian epithelium. Sequence analysis of these alternative transcripts determined that they derived from PARK2and contained exonic duplications and/or deletions. The same research group later confirmed these results and investigated the expression of parkin in seven primary ovarian tumors and six ovarian tumor-derived cell lines by Western blot analysis. Results showed that some of these samples exhibit decreased or the complete absence of Parkin protein expression [57].
CONCLUSIONS
AS is a complex molecular mechanism, which increases the coding capacity of our genome by generating distinct proteins from a single gene. The importance of AS in the regulation of diverse biological processes is mirrored in the growing list of human diseases associated with known or suspected splicing defects, including neurodegenerative and oncologic diseases. A great number of evidences support the intricate and complex AS regulating the expression of PARK2 gene. Mutations that change this delicate equilibrium can lead to alterations in the levels of the correctly spliced forms, or can alter their physiological role. Investigating the distribution of PARK2 splice forms, their functional characterization and the AS-altering mutations could open up new scenarios for the resolution of some molecular ways contributing to human diseases.
ACKNOWLEDGEMENTS
This work was supported by the Italian Ministry of Education, Universities and Research through grant CTN01_00177_817708 and the international Ph.D. program in Neuroscience of the University of Catania. Authors gratefully acknowledge Cristina Calì, Alfia Corsino, Maria Patrizia D’Angelo and Francesco Marino for their administrative and technical support.
REFERENCES
- Pan, Q.; Shai, O.; Lee, L. J.; Frey, B. J.; Blencowe, B. J., Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 2008,40 (12), 1413-5.
- Wang, E. T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S. F.; Schroth, G. P.; Burge, C. B., Alternative isoform regulation in human tissue transcriptomes. Nature 2008,456 (7221), 470-6.
- Yap, K.; Makeyev, E. V., Regulation of gene expression in mammalian nervous system through alternative pre-mRNA splicing coupled with RNA quality control mechanisms. Molecular and cellular neurosciences 2013,56, 420-8.
- La Cognata, V.; D'Agata, V.; Cavalcanti, F.; Cavallaro, S., Splicing: is there an alternative contribution to Parkinson's disease? Neurogenetics 2015.
- Faustino, N. A.; Cooper, T. A., Pre-mRNA splicing and human disease. Genes & development 2003,17 (4), 419-37.
- Ward, A. J.; Cooper, T. A., The pathobiology of splicing. The Journal of pathology 2010,220 (2), 152-63.
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N., Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998,392 (6676), 605-8.
- Matsumine, H.; Saito, M.; Shimoda-Matsubayashi, S.; Tanaka, H.; Ishikawa, A.; Nakagawa-Hattori, Y.; Yokochi, M.; Kobayashi, T.; Igarashi, S.; Takano, H.; Sanpei, K.; Koike, R.; Mori, H.; Kondo, T.; Mizutani, Y.; Schaffer, A. A.; Yamamura, Y.; Nakamura, S.; Kuzuhara, S.; Tsuji, S.; Mizuno, Y., Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2-27. Am J Hum Genet 1997,60 (3), 588-96.
- Ikeuchi, K.; Marusawa, H.; Fujiwara, M.; Matsumoto, Y.; Endo, Y.; Watanabe, T.; Iwai, A.; Sakai, Y.; Takahashi, R.; Chiba, T., Attenuation of proteolysis-mediated cyclin E regulation by alternatively spliced Parkin in human colorectal cancers. Int J Cancer 2009,125 (9), 2029-35.
- Kitada, T.; Asakawa, S.; Minoshima, S.; Mizuno, Y.; Shimizu, N., Molecular cloning, gene expression, and identification of a splicing variant of the mouse parkin gene. Mamm Genome 2000,11 (6), 417-21.
- Beyer, K.; Domingo-Sabat, M.; Humbert, J.; Carrato, C.; Ferrer, I.; Ariza, A., Differential expression of alpha-synuclein, parkin, and synphilin-1 isoforms in Lewy body disease. Neurogenetics 2008,9 (3), 163-72.
- Humbert, J.; Beyer, K.; Carrato, C.; Mate, J. L.; Ferrer, I.; Ariza, A., Parkin and synphilin-1 isoform expression changes in Lewy body diseases. Neurobiol Dis 2007,26 (3), 681-7.
- Tan, E. K.; Shen, H.; Tan, J. M.; Lim, K. L.; Fook-Chong, S.; Hu, W. P.; Paterson, M. C.; Chandran, V. R.; Yew, K.; Tan, C.; Yuen, Y.; Pavanni, R.; Wong, M. C.; Puvan, K.; Zhao, Y., Differential expression of splice variant and wild-type parkin in sporadic Parkinson's disease. Neurogenetics 2005,6 (4), 179-84.
- Dagata, V.; Cavallaro, S., Parkin transcript variants in rat and human brain. Neurochemical research 2004,29 (9), 1715-24.
- Sunada, Y.; Saito, F.; Matsumura, K.; Shimizu, T., Differential expression of the parkin gene in the human brain and peripheral leukocytes. Neuroscience letters 1998,254 (3), 180-2.
- Huynh, D. P.; Scoles, D. R.; Ho, T. H.; Del Bigio, M. R.; Pulst, S. M., Parkin is associated with actin filaments in neuronal and nonneural cells. Annals of neurology 2000,48 (5), 737-44.
- Horowitz, J. M.; Myers, J.; Stachowiak, M. K.; Torres, G., Identification and distribution of Parkin in rat brain. Neuroreport 1999,10 (16), 3393-7.
- D'Agata, V.; Grimaldi, M.; Pascale, A.; Cavallaro, S., Regional and cellular expression of the parkin gene in the rat cerebral cortex. The European journal of neuroscience 2000,12 (10), 3583-8.
- La Cognata, V.; Iemmolo, R.; D'Agata, V.; Scuderi, S.; Drago, F.; Zappia, M.; Cavallaro, S., Increasing the Coding Potential of Genomes Through Alternative Splicing: The Case of PARK2 Gene. Current genomics 2014,15 (3), 203-16.
- Scuderi, S.; La Cognata, V.; Drago, F.; Cavallaro, S.; D'Agata, V., Alternative splicing generates different parkin protein isoforms: evidences in human, rat, and mouse brain. BioMed research international 2014,2014, 690796.
- D'Amico, A. G.; Maugeri, G.; Magro, G.; Salvatorelli, L.; Drago, F.; D'Agata, V., Expression pattern of parkin isoforms in lung adenocarcinomas. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine 2015,36 (7), 5133-41.
- Maugeri, G.; D'Amico, A. G.; Magro, G.; Salvatorelli, L.; Barbagallo, G. M.; Saccone, S.; Drago, F.; Cavallaro, S.; D'Agata, V., Expression profile of parkin isoforms in human gliomas. International journal of oncology 2015,47 (4), 1282-92.
- Ambroziak, W.; Koziorowski, D.; Duszyc, K.; Gorka-Skoczylas, P.; Potulska-Chromik, A.; Slawek, J.; Hoffman-Zacharska, D., Genomic instability in the PARK2 locus is associated with Parkinson's disease. Journal of applied genetics 2015,56 (4), 451-61.
- West, A. B.; Lockhart, P. J.; O'Farell, C.; Farrer, M. J., Identification of a novel gene linked to parkin via a bi-directional promoter. Journal of molecular biology 2003,326 (1), 11-9.
- D'Agata, V.; Zhao, W.; Pascale, A.; Zohar, O.; Scapagnini, G.; Cavallaro, S., Distribution of parkin in the adult rat brain. Progress in neuro-psychopharmacology & biological psychiatry 2002,26 (3), 519-27.
- Seirafi, M.; Kozlov, G.; Gehring, K., Parkin structure and function. The FEBS journal 2015,282 (11), 2076-88.
- Imai, Y.; Soda, M.; Takahashi, R., Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. The Journal of biological chemistry 2000,275 (46), 35661-4.
- Shimura, H.; Hattori, N.; Kubo, S.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; Suzuki, T., Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 2000,25 (3), 302-5.
- Staropoli, J. F.; McDermott, C.; Martinat, C.; Schulman, B.; Demireva, E.; Abeliovich, A., Parkin is a component of an SCF-like ubiquitin ligase complex and protects postmitotic neurons from kainate excitotoxicity. Neuron 2003,37 (5), 735-49.
- Chung, K. K.; Zhang, Y.; Lim, K. L.; Tanaka, Y.; Huang, H.; Gao, J.; Ross, C. A.; Dawson, V. L.; Dawson, T. M., Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nature medicine 2001,7 (10), 1144-50.
- Zhang, Y.; Gao, J.; Chung, K. K.; Huang, H.; Dawson, V. L.; Dawson, T. M., Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proceedings of the National Academy of Sciences of the United States of America 2000,97 (24), 13354-9.
- Chan, N. C.; Salazar, A. M.; Pham, A. H.; Sweredoski, M. J.; Kolawa, N. J.; Graham, R. L.; Hess, S.; Chan, D. C., Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Human molecular genetics 2011,20 (9), 1726-37.
- Yoshii, S. R.; Kishi, C.; Ishihara, N.; Mizushima, N., Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. The Journal of biological chemistry 2011,286 (22), 19630-40.
- Cookson, M. R., Parkinsonism due to mutations in PINK1, parkin, and DJ-1 and oxidative stress and mitochondrial pathways. Cold Spring Harbor perspectives in medicine 2012,2 (9), a009415.
- Jin, S. M.; Youle, R. J., PINK1- and Parkin-mediated mitophagy at a glance. Journal of cell science 2012,125 (Pt 4), 795-9.
- Narendra, D.; Tanaka, A.; Suen, D. F.; Youle, R. J., Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. The Journal of cell biology 2008,183 (5), 795-803.
- Kasap, M.; Akpinar, G.; Sazci, A.; Idrisoglu, H. A.; Vahaboglu, H., Evidence for the presence of full-length PARK2 mRNA and Parkin protein in human blood. Neuroscience letters 2009,460 (3), 196-200.
- Nuytemans, K.; Theuns, J.; Cruts, M.; Van Broeckhoven, C., Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Human mutation 2010,31 (7), 763-80.
- Bonifati, V., Autosomal recessive parkinsonism. Parkinsonism & related disorders 2012,18 Suppl 1, S4-6.
- Lucking, C. B.; Durr, A.; Bonifati, V.; Vaughan, J.; De Michele, G.; Gasser, T.; Harhangi, B. S.; Meco, G.; Denefle, P.; Wood, N. W.; Agid, Y.; Brice, A.; French Parkinson's Disease Genetics Study, G.; European Consortium on Genetic Susceptibility in Parkinson's, D., Association between early-onset Parkinson's disease and mutations in the parkin gene. N Engl J Med 2000,342 (21), 1560-7.
- Oliveira, S. A.; Scott, W. K.; Martin, E. R.; Nance, M. A.; Watts, R. L.; Hubble, J. P.; Koller, W. C.; Pahwa, R.; Stern, M. B.; Hiner, B. C.; Ondo, W. G.; Allen, F. H., Jr.; Scott, B. L.; Goetz, C. G.; Small, G. W.; Mastaglia, F.; Stajich, J. M.; Zhang, F.; Booze, M. W.; Winn, M. P.; Middleton, L. T.; Haines, J. L.; Pericak-Vance, M. A.; Vance, J. M., Parkin mutations and susceptibility alleles in late-onset Parkinson's disease. Ann Neurol 2003,53 (5), 624-9.
- Illarioshkin, S. N.; Periquet, M.; Rawal, N.; Lucking, C. B.; Zagorovskaya, T. B.; Slominsky, P. A.; Miloserdova, O. V.; Markova, E. D.; Limborska, S. A.; Ivanova-Smolenskaya, I. A.; Brice, A., Mutation analysis of the parkin gene in Russian families with autosomal recessive juvenile parkinsonism. Mov Disord 2003,18 (8), 914-9.
- Pigullo, S.; De Luca, A.; Barone, P.; Marchese, R.; Bellone, E.; Colosimo, A.; Scaglione, C.; Martinelli, P.; Di Maria, E.; Pizzuti, A.; Abbruzzese, G.; Dallapiccola, B.; Ajmar, F.; Mandich, P., Mutational analysis of parkin gene by denaturing high-performance liquid chromatography (DHPLC) in essential tremor. Parkinsonism & related disorders 2004,10 (6), 357-62.
- Scherfler, C.; Khan, N. L.; Pavese, N.; Eunson, L.; Graham, E.; Lees, A. J.; Quinn, N. P.; Wood, N. W.; Brooks, D. J.; Piccini, P. P., Striatal and cortical pre- and postsynaptic dopaminergic dysfunction in sporadic parkin-linked parkinsonism. Brain 2004,127 (Pt 6), 1332-42.
- Bertoli-Avella, A. M.; Giroud-Benitez, J. L.; Akyol, A.; Barbosa, E.; Schaap, O.; van der Linde, H. C.; Martignoni, E.; Lopiano, L.; Lamberti, P.; Fincati, E.; Antonini, A.; Stocchi, F.; Montagna, P.; Squitieri, F.; Marini, P.; Abbruzzese, G.; Fabbrini, G.; Marconi, R.; Dalla Libera, A.; Trianni, G.; Guidi, M.; De Gaetano, A.; Boff Maegawa, G.; De Leo, A.; Gallai, V.; de Rosa, G.; Vanacore, N.; Meco, G.; van Duijn, C. M.; Oostra, B. A.; Heutink, P.; Bonifati, V.; Italian Parkinson Genetics, N., Novel parkin mutations detected in patients with early-onset Parkinson's disease. Mov Disord 2005,20 (4), 424-31.
- Bardien, S.; Keyser, R.; Yako, Y.; Lombard, D.; Carr, J., Molecular analysis of the parkin gene in South African patients diagnosed with Parkinson's disease. Parkinsonism & related disorders 2009,15 (2), 116-21.
- Nuytemans, K.; Meeus, B.; Crosiers, D.; Brouwers, N.; Goossens, D.; Engelborghs, S.; Pals, P.; Pickut, B.; Van den Broeck, M.; Corsmit, E.; Cras, P.; De Deyn, P. P.; Del-Favero, J.; Van Broeckhoven, C.; Theuns, J., Relative contribution of simple mutations vs. copy number variations in five Parkinson disease genes in the Belgian population. Hum Mutat 2009,30 (7), 1054-61.
- Pankratz, N.; Kissell, D. K.; Pauciulo, M. W.; Halter, C. A.; Rudolph, A.; Pfeiffer, R. F.; Marder, K. S.; Foroud, T.; Nichols, W. C.; Parkinson Study Group, P. I., Parkin dosage mutations have greater pathogenicity in familial PD than simple sequence mutations. Neurology 2009,73 (4), 279-86.
- Brudek, T.; Winge, K.; Bredo Rasmussen, N.; Bahl Czarna, J. M.; Tanassi, J.; Klitmoller Agander, T.; Hyde, T. M.; Pakkenberg, B., Altered Alpha-Synuclein, Parkin, and Synphilin Isoform Levels in Multiple System Atrophy Brains. Journal of neurochemistry 2015.
- Cesari, R.; Martin, E. S.; Calin, G. A.; Pentimalli, F.; Bichi, R.; McAdams, H.; Trapasso, F.; Drusco, A.; Shimizu, M.; Masciullo, V.; D'Andrilli, G.; Scambia, G.; Picchio, M. C.; Alder, H.; Godwin, A. K.; Croce, C. M., Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27. Proceedings of the National Academy of Sciences of the United States of America 2003,100 (10), 5956-61.
- Iwakawa, R.; Okayama, H.; Kohno, T.; Sato-Otsubo, A.; Ogawa, S.; Yokota, J., Contribution of germline mutations to PARK2 gene inactivation in lung adenocarcinoma. Genes, chromosomes & cancer 2012,51 (5), 462-72.ARC Journal of Neuroscience Page | 18
- Xiong, D.; Wang, Y.; Kupert, E.; Simpson, C.; Pinney, S. M.; Gaba, C. R.; Mandal, D.; Schwartz, A. G.; Yang, P.; de Andrade, M.; Pikielny, C.; Byun, J.; Li, Y.; Stambolian, D.; Spitz, M. R.; Liu, Y.; Amos, C. I.; Bailey-Wilson, J. E.; Anderson, M.; You, M., A recurrent mutation in PARK2 is associated with familial lung cancer. American journal of human genetics 2015,96 (2), 301-8.
- Veeriah, S.; Taylor, B. S.; Meng, S.; Fang, F.; Yilmaz, E.; Vivanco, I.; Janakiraman, M.; Schultz, N.; Hanrahan, A. J.; Pao, W.; Ladanyi, M.; Sander, C.; Heguy, A.; Holland, E. C.; Paty, P. B.; Mischel, P. S.; Liau, L.; Cloughesy, T. F.; Mellinghoff, I. K.; Solit, D. B.; Chan, T. A., Somatic mutations of the Parkinson's disease-associated gene PARK2 in glioblastoma and other human malignancies. Nature genetics 2010,42 (1), 77-82.
- Yeo, C. W.; Ng, F. S.; Chai, C.; Tan, J. M.; Koh, G. R.; Chong, Y. K.; Koh, L. W.; Foong, C. S.; Sandanaraj, E.; Holbrook, J. D.; Ang, B. T.; Takahashi, R.; Tang, C.; Lim, K. L., Parkin pathway activation mitigates glioma cell proliferation and predicts patient survival. Cancer research 2012,72 (10), 2543-53.
- Lin, D. C.; Xu, L.; Chen, Y.; Yan, H.; Hazawa, M.; Doan, N.; Said, J. W.; Ding, L. W.; Liu, L. Z.; Yang, H.; Yu, S.; Kahn, M.; Yin, D.; Koeffler, H. P., Genomic and Functional Analysis of the E3 Ligase PARK2 in Glioma. Cancer research 2015,75 (9), 1815-27.
- Denison, S. R.; Callahan, G.; Becker, N. A.; Phillips, L. A.; Smith, D. I., Characterization of FRA6E and its potential role in autosomal recessive juvenile parkinsonism and ovarian cancer. Genes, chromosomes & cancer 2003,38 (1), 40-52.
- Denison, S. R.; Wang, F.; Becker, N. A.; Schule, B.; Kock, N.; Phillips, L. A.; Klein, C.; Smith, D. I., Alterations in the common fragile site gene Parkin in ovarian and other cancers. Oncogene 2003,22 (51), 8370-8.