


|
|
DOI Prefix 10.20431 |
Information
Journal Policies
Collagenofibrotic Glomerulopathy -A Rare Entity
Dhanapriya.J1*,Dinesh kumar.T1,Arcot Thanjan1,Anila Abraham kurien1,Sakthirajan.Ramanathan1,T.Balasubramaniyan1,N.Gopalakrishnan1
Copyright : © 2017 . This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Collagenofibrotic glomerulopathy (CG) is a rare type of deposit glomerulopathies characterized by deposits of 60–80 nm fibrils in the sub-endothelial and mesangial areas and increased levels of serum pro-collagen type III peptide (PIIINP). Fewer than 50 cases have been reported in the literature, mostly from Japan. We report a 21-year old female patient who presented with nephrotic syndrome and renal biopsy showed nodular glomerulosclerosis by light microscopy (LM) and electromicroscopy (EM) revealed frayed curvilinear collagen fibers confirming diagnosis of collagenofibrotic glomerulopathy . CG is a rare differential diagnosis of nodular glomerulosclerosis diagnosed with the help of EM. No specific therapy is available.
Collagenofibrotic glomerulopathy, electron microscopy fibrils, type III collagen glomerulopathy,Nephrology
1. Introduction
Collagenofibrotic glomerulopathy may present as an isolated, sporadic form usually seen in adults, or as a familial form with autosomal recessive inheritance, seen in children.[1] CG was first described in 1979 from Japan by Masaaki and Arakawa and published a series of ten cases [2].We describe here a female patient with collagenofibrotic glomerulopathy.
2. Case Report
A 21-year-old female, presented with edema legs on and off for the past 3 years duration. There was no connective tissue disease or family history of renal disease. On examination, patient was anemic and normotensive. Laboratory investigations showed urinalysis:3+ proteinuria and bland urine sediment ; 24-hour proteinuria:5.1 gm; blood haemoglobin:4.5 gm ; total leucocyte count:8000/cu.mm; platelet count:3.2 lakh/cu.mm; blood urea:38 mg / dl ; serum creatinine:2.2 mg/dl; corrected serum calcium:8.9 mg/dl ;serum phosphorous :4.8 mg/dl ; serum ANA: negative; serum C3:97mg/dl; serum C4:15 mg/dl. Ultrasonogram and Computed tomography scan of the abdomen was normal. Serological tests for hepatitis B, hepatitis C, Ebstein Barr virus and HIV were negative. Serum protein electrophoresis, serum immunofixation, free light chain assay and bone marrow examination were negative for paraproteinemias. Serum PIIINP levels and collagen profile could not be estimated due to logistic reasons.
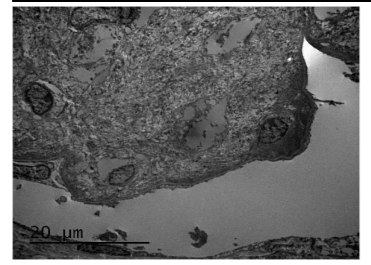
Renal Biopsy revealed enlarged glomeruli with diffuse and nodular mesangial expansion which PAS and congo red negative deposits but stains blue on trichrome stain and was equivocal on silver stain. Interstitial fibrosis and tubular atrophy involved about 20-25% of the core. Immunofluorescence (IF) was negative for IgG, IgM, IgA, C3, C1q, Kappa& Lambda light chains. Ultrastructural analysis showed massive deposition of numerous electron dense curvilinear and banded fibrils of size 60 nm with splayed ends( aberrant collagen )within the mesangium and along glomerular capillary loops consistent with CG(figure1). Patient initiated on antiproteinuric measures (ACE inhibitors and statins).On follow up after 6 months, serum creatinine was: 2.2mg/dl and had partial remission of proteinuria.
Figure1a. Renal biopsy showing nodular mesangial expansion (trichrome)
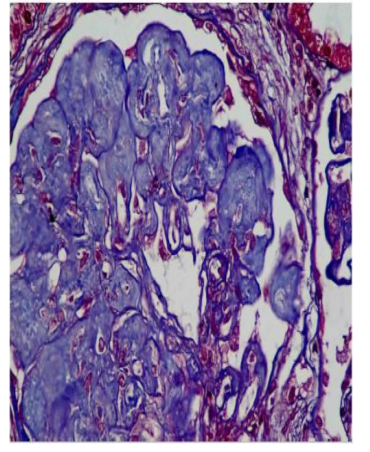
Figure1b. Electron microscopy image showing massive deposition of curvilinear aberrant collagen fibrils in mesangium(12000 X)
3. Discussion
Collagenofibrotic glomerulopathy is a disorder of unknown etiology often occurs sporadically in adults, with no age or sex predilection. [3] In 1993, Gubler et al from France, reported 10 children of this disorder, who had familial autosomal recessive inheritance. [4] Clustering of the cases from Japan points to environmental or racial factors. Clinical presentation includes nephrotic proteinuria (60%), hypertension (66%) and anemia. [5] Renal function test results usually are normal or slightly increased at presentation.
In the human kidney, type III collagen is present only in the interstitium and blood vessels and not in the glomerulus. There are two concepts regarding the origin of the accumulated aberrant collagen.[6] One is that the abnormal type III collagen is produced endogenously by the mesangium. Interleukin 4 (IL-4) selectively stimulated type III collagen synthesis in human glomerular cells and IL-4 – neutralizing antibodies have prevented renal disease in IL-4 transgenic mice, which developed glomerulosclerosis in the absence of immunoglobulin deposition. [6,7] Other shows some evidence of extra-renal involvement leading to a hypothesis that it may be a systemic disease with abnormal metabolism of Type III collagen. Increased circulating levels of PIIINP, an autopsy report of collagen III deposition in all organs and association of hepatic perisinusoidal fibrosis in a case of CG suggest the possibility of systemic disorder. Kopp et al have described a transgenic mouse model that develops glomerular lesions similar to human collagenofibrotic glomerulopathy involving active form of TGF- B1[7].
Renal biopsy reveals PAS and congo red stain negative but aniline blue and acid fuschin orange positive eosinophilic sub-endothelial and mesangial deposits.IF for immunoglobins, complement components and light chains is negative and characteristically positive for type III collagen. EM shows marked accumulation of irregularly arranged bundles of fibrillar material of size 50 to 60 nm in the mesangium and sub-endothelial space.[8] In our patient, nodular glomeruli prompted the differential diagnosis of membranoproliferative glomerulonephritis type I, chronic thrombotic microangiopathy, amyloidosis,diabetic glomerulo-sclerosis, and light chain deposition disease but was ruled out. Abnormal accumulation of banded collagen predominantly within the lamina densa of glomerular basement membranes with lucent areas (moth-eaten appearance) in EM is the hallmark of nail-patella syndrome glomerulopathy where as, in CG the abnormal collagen bundles are predominantly in the mesangium and subendothelial region[9].
Serum PIIINP levels will be elevated markedly (10-100 times normal), in CG. Immunostaining with fluorescein-conjugated antibodies to collagen type III in the mesangium and capillary walls can confirm the diagnosis, but it was not performed in this case. Kurien et al from India described first case series in adults. The average age was 38 years with five males and three females. [10] All patients presented with nephrotic syndrome, and five had hypertension. The average serum creatinine was 146.5 μmol/L (88.4–282.9 μmol/L range).
No specific therapy is available. Steroid therapy is reported to be effective in temporarily treating the renal dysfunction, anemia and proteinuria. The natural history is variable, but the disease is progressive, with progression to end-stage renal disease in 5–12 years. Successful kidney transplantation has been reported with no recurrence at three years.
4. Conclusion
Though the occurrence of CG is rare, definite diagnosis can be established when typical histological findings .The natural history of the disease is variable and has no specific treatment. The etiology and pathogenesis remain elusive.
References
- Alchi B, Nishi S, Narita I, Gejyo F. Collagenofibrotic glomerulopathy: clinicopathologic overview of a rare glomerular disease. Am J Kidney Dis 2007;49:499–506.
- Arakawa M. Idiopathic mesangio-degenerative glomerulopathy. Jpn J Nephrol 1979; 21: 914– 5.
- Patro KC, Jha R, Sahay M , Swarnalatha G.Collagenofibrotic glomerulopathy- case report with review of literature. Indian J Nephrol 2011; 21: 52–5.
- Gubler MC, Dommergues JP, Foulard M, et al. Collagen type III glomerulopathy: a new type of hereditary nephropathy. Pediatr Nephrol 1993; 7:354–60.
- Duggal R, Nada R, Rayat CS, Rane SU, Sakhuja V, Joshi K. Collagenofibrotic glomerulopathy-a review. Clin Kidney J. 2012; 5:7-12.
- Yasuda T, Imai H, Nakamoto Y, Ohtani H, Komatsuda A, Wakui H, et al. Collagenofibrotic glomerulopathy: a systemic disease. Am J Kidney Dis 1999; 33: 123–7.
- Kopp JB, Factor VM, Mozes M, Nagy P, Sanderson N, Böttinger EP, et al. Transgenic mice with increased plasma levels of TGF-beta 1 develop progressive renal disease. Lab Invest 1996; 74:991–1003.
- Soni SS, Gowrishankar S, Nagarik AP, Barnela SR, De Padua M, Adikey GK, et al. Collagenofibrotic glomerulopathy in association with hodgkin’s lymphoma.Saudi J Kidney Dis Transpl 2011; 22: 126–9.
- Sabnis SG, Antonovych TT, Argy WP, Rakowski TA, Gandy DR, Salcedo JR. Nail– Patella syndrome.Clin Neph 1980; 14:148–53.
- Kurien A A,Larsen C P ,Cossey LN . Collagenofibrotic glomerulopathy.Clin Kidney J. 2015; 8: 543–7.