


|
|
DOI Prefix 10.20431 |
Information
Journal Policies
Study of Aplastic Anaemia, Etiological Role of Environmental and Genetic Factors in Eastern India-A Review
Atreyee Dutta M.Sc1, Dr. Rajib De D.M2, Dr. Tuphan Kanti Dolai D.M3, Dr. Pradip Kumar Mitra M.D4, Dr. Ajanta Halder Ph.D5*
2.Associate Professor, Department of Haematology, Nil Ratan Sircar Medical College and Hospital, 138, AJC Bose Road, Kolkata-700014, West Bengal, India.
3.Professor, Department of Haematology, Nil Ratan Sircar Medical College and Hospital138, AJC Bose Road, Kolkata-70014, West Bengal, India.
4.Director of Medical Education, Department Of Health, Government of West Bengal, GN-29, GN Block, Sector V, Kolkata-700091, West Bengal, India.
5.Associate Professor, Department of Genetics, Vivekananda Institute of Medical Sciences, Ramakrishna Mission Seva Pratishthan, 99 Sarat Bose Road, Kolkata-700026, West Bengal, India.
Copyright : © 2018 Authors. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Aplastic anaemia, an uncommon hematologic complication, arises from the incidence of bone marrow failure. Universally it has been acknowledged as a deadly blood disorder. Fortunately few years back some specific treatment has been evolved. With the upgradation of the existing knowledge, it is known that stem-cell transplantation and immunosuppressive drug therapy are the choice of treatments of aplastic anaemia. Aplastic anaemia can be inherited but in most of the patients pathophysiology is immune mediated and is called acquired aplastic anaemia. The activated cytotoxic T-lymphocytes mediated dystruction of hematopoietic stem cell is the basis of the pathophysiological mechanism. The majority of patient responds to the immunosuppression with anti-thymocyte globulins and cyclosporine by restoring blood-cell production but still the relapse of the disease and especially evolution of clonal hematologic diseases remain a challenge. In the young patients allogeneic stem-cell transplant from histocompatible sibling donors is the best curative treatment. But many patients do not have histocompatible sibling donors. Especially in the older patients, transplantation is not fruitful. Nowadays alternative source of stem cells and a variety of conditioning regimens are in the spot light of research and the results have shown a promising engraftment, competing with conventional transplantation. In this first review article on aplastic anaemia from eastern India, we tried to enunciate the etiological role of environmental and genetic factors associated with aplastic anaemia. This review will inculcate the precise understanding of the disease to its readers.
Aplastic anaemia, Abnormal cytogenetic clone, Stress cytogenetics, Cytokines, Apoptosis, Environmental factors, Genetic factors, Idiopathic, Haematopoietic stem cell transplantation, Immunosuppressive therapy ,Hematology
1. Introduction
By origin, aplastic anaemia (AA) was considered to be heterogeneous and thereby was virtually impossible to study methodically. In due course of time, as progenitor assays were established, different factors could be held supposedly accountable for failure to form colonies in tissue culture, ranging from quantitative and qualitative flaws in haematopoietic stem cells during differentiation to a lack of stroma support or inadequate cytokine production or the effects of a chemical poison. In superseding periods, with advancement in research, our understanding of AA has stuck around a combined immune mechanism of haematopoietic stem cells damage. Remission was achieved in the AA patients after effective immunosuppressive therapies. Nowadays, with the developments of technical knowledge few numbers of cells can be effortlessly assayed by cell biology, biotechnology,flow-cytometry,molecular biology and immunology. As a result, we have got a more depth, lucid, sensible and rational view of the pathophysiology of AA. Now it can be stated that, AA is a rare but heterogeneous haematological disorder characterized by pancytopenia with a hypocellular bone marrow. Bone marrow (BM) is a soft, spongy tissue. This is the factory which makes enough red blood cells (RBC), white blood cells (WBC) and platelets. The red cells carry O2, CO2 to the body, white cells that fight against infection, and platelets helps in blood coagulation. BM failure happens when it is unable to keep up with the body's need for healthy blood cells. In approximately 15-20% of patients the disease is constitutional or inherited (IAA- inherited aplastic anaemia)[1]. Despite advancement in our understanding of the pathophysiology of AA, still 70-80% of these patients are categorized as acquired aplastic anaemia (AAA). It has also been documented that in 50% of the confirmed patients of AAA, the cause of the disease is unknown[2]. It is called idiopathic aplastic anaemia when the primary aetiology is unknown. A global forecast report on AAA market research is expecting to grow at a CAGR (compound annual growth rate) of 7.6 % during the forecast period 2017-2023. AA shows a bimodal distribution with highest frequency occurring between the ages of 15-25 years and a second peak after 60 years[3]. Being an Asian descent makes someone 3 times vulnerable to develop this disease at any point of their life than a European. It can happen to anyone of any race and ethnicity. This disease affects men at the same rates that it affects women. The treatment of AA is performed by allogeneic stem-cell transplantation or immunosuppressive therapy (IST). It is understood that the disease AA share an overlapping field with some other marrow failure syndromes. Clonal hematopoiesis in AA has been found to be linked to the evolution of late clonal disorders, including paroxysmal nocturnal haemoglobinuria (PNH), myelo-dysplastic syndromes (MDS) and acute myeloid leukaemia (AML). These are the common complications after successful immunosuppressive therapy (IST). In some autoimmune diseases where the other organ systems are involved, an occasional marrow aplasia can be pointed out. Our concern in this constrained review is to highlight the etiological role of environmental and genetic factors associated with AA. This is the first review article on AA from Eastern India. This will be helpful for the readers to achieve an insightful knowledge on AA.
2. Etiological Classification
Depending on the contemporary knowledge AA can be divided in to two categories[3]
i. Congenital/Hereditary/Genetic/Inherited Aplastic Anaemia
ii. Acquired aplastic anaemia
Fanconi Anaemia (FA) is the mostly found constitutional, an autosomal recessive AA. It is discovered by Guido Fanconi in three brothers’ in 1927[4]. FA results from the defects in genes that is responsible for the modulation of DNA stability. It is a rare disorder with incidence of 1 in 360,000 births[5]. It is frequent in Afrikaners who are European descent[6].
Dyskeratosis Congenita (DKC), also known as Zinsser-Engman-Cole syndrome, was first described in 1906. Evidence exists for telomerase dysfunction, ribosome deficiency, and protein synthesis dysfunction in this disorder. It is much more common in males than in females and occurs in approximately 1 per million population[7].
Shwachman-diamond syndrome (SDS) was first described in 1964[8]. It is an uncommon inherited disorder that found in every 100,000 births. It affects 1 in 75000 AA patients[9].
The disease was discovered by Hugh W. Josephs in 1936[10]. The condition is however named after the pediatricians Louis K. Diamond Kenneth Blackfan, who described congenital hypoplastic anaemia in 1938[11]. Diamond-Blackfan anaemia (DBA) is caused by mutations in several genes. Identified genes include but are not limited to: RPS19, RPL5, RPS10, RPL11, RPL35A, RPS7, RPS17, RPS24, RPS26 and GATA1 genes. The incidence of DBA is 1:100,000 and 1:200,000 live births; it remains consistent across ethnicities[12].
Several other rare syndromes are associated with aplastic pancytopenia. These are Ataxia-pancytopenia (myelocerebellar disorder),[13] Congenital thrombocytopenia,[14] DNA ligase IV deficiency,[15] Dubowitz syndrome,[16] Nijmegen breakage syndrome,[17] Reticular dysgenesis[18] Seckel syndrome,[19] and WT syndrome[20].
In acquired aplastic anaemia (AAA), the causesitive factor can be drugs, toxic chemicals, viruses, Paroxymal Nocturnal Hemoglobinurea, Autoimmune/connective tissue disorders, Thymoma and thymic carcinoma, Pregnancy, Iatrogenic and sometimes unknown factors.
Previously only few drugs were thought to be related with AAA but from various research and studies,[21] now the drugs can be divided into three categories 1) High risk, 2) Intermediate Risk and 3) Low Risk. Surprisingly, no reasonable mechanism has been established for the most infamous pharmaceutical, chloramphenicol, or for other agents such as penicillamine or gold. Patients of permanent marrow aplasia after idiosyncratic exposure must be studied vigorously to illuminate the extremely sensitive mechanism of the hidden metabolites.
The first chemical was benzene, found to be associated with AA[22]. Benzene is a widely used chemical and involved in the manufacture of chemicals, drugs, dyes, explosives, rubber, leather goods, shoe industry etc… Many patients of AA have been attributed to pentachlorophenol (PCP) over the past 25 years[23]. Prolonged exposures to petroleum (Stoddard solvent, smoke, liquid form) can be a cause of marrow aplasia[24]. Again toluene is an extensively used industrial chemical. Trinitrotoluene (TNT), an explosive, is absorbed readily by inhalation and through the skin[25].
Many workers exposed to TNT in Great Britain during 1940-1946 were in the claws of AA[26].
Even the acute exposure to toluene through the practice of glue sniffing also have been described to cause marrow aplasia[27]. Some studies have focused that pesticides (Organochlorine and Organophosphate) are related with the onset [23] to the disease, especially those who are associated with agricultural exposure[28]. DDT (dichlorodiphenyltrichloroethane), lindane and chlordane are insecticides which are linked to AA[23,29].
Hepatitis, following the development of AA has been the observed, and this association was clearly highlighted by the two major reviews[30]. Although hepatitis A and B have been implicated in aplastic anaemia in a small number of patients, but most of the patients, are linked to non-A, non-B, non-C hepatitis[31]. many reports recommend a relationship of parvovirus B19 with aplastic anaemia,[32] whereas others have not[33]. Epstein-Barr virus (EBV) has been found to be associated with pathogenesis of aplastic anaemia[34]. Human immunodeficiency virus (HIV) infection is often associated with cytopenia. The marrow is occasionally hypoplastic[35]. Human herpes virus (HHV)-6 is a cause of severe marrow aplasia in post transplantation condition.[36]
PNH is characterized by an acquired mutation in the PIG-A gene that encodes an enzyme that is required to synthesize mannolipids. Deficiency of mannolipids prevents the synthesis of the glycosyl-phosphatidylinositol anchor precursor. This moiety anchors several proteins, including inhibitors of the complement pathway to blood cell membranes and its absence accounts for the complement-mediated hemolysis in PNH. PNH is often associated with reduced BM function caused by AA[37].
The systemic lupus erythematosus is irregularly associated with AA[38]. The patients have improved after plasmapheresis,[39] glucocorticoids [40] or cyclophosphamide therapy [41]which is consistent with an immune etiology. Eosinophilic fasciitis, a rare connective tissue disorder has been associated with aplastic anaemia. The manifestation includes painful swelling, induration of the skin and subcutaneous tissue[42]. Auto immune thyroid diseases are reported to be associated with AA[43]. AA has also been reported in association with thymoma[40]. The causal connection may be the role of cytotoxic T lymphocytes in the pathogenesis of numerous autoimmune diseases and in AA[44].
Ehrlich describes the first incidence of AA in a pregnant woman[45]. There are a number of reports of pregnancy-associated aplastic anaemia, but the connection between the two circumstances is unclear[46,47]. In some patients, pre-existing aplastic anaemia is aggravated with pregnancy, recovery achieved following termination of the pregnancy. A woman patient of AA previously treated with immunosuppression can give birth of a normal new born[47].
Radiation: A dose of 4.5 Gy (Gray) leads to death in half the individuals (LD50) and higher doses in the range of 10 Gy are universally fatal.
Cytotoxic drug therapy: BM aplasia follows cytotoxic drug therapy for cancers and is an anticipated and transient toxic effect for a variety of nonmalignant diseases.
Very few studies have emphasized on the stromal microenvironment and growth factors. In AA patients serum levels of stem cell factor (SCF) have been found either moderately low or normal[48]. Another growth factor, FLT-3 ligand, is 30 to 100-fold higher in the serum of patients with aplastic anaemia[49]. Elevated level of granulocyte colony-stimulating factor,[50] erythropoietin,[51] and thrombopoietin (TPO) [52] are found in AA. IL-1, an early stimulator of hematopoiesis, is decreased in mononuclear cells from patients with AA[53]. Microenvironment studies have shown relatively normal stromal cell proliferation and growth factor construction[54].
Genetic dissimilarities in drug metabolism, particularly in detoxification of reactive intermediate compounds is still not in attention. Some studies have focused on the over representation or deletions in the drug-metabolizing glutathione-S-transferase genes GSTM1, GSTT1, which would increase concentrations of toxic drug intermediates[55].
For transfusional support leukocyte-depleted blood product should be used to minimise the chance of leukocyte & platelet sensitization, transfusion reactions and graft-versus-host disease (GVHD). It is important not to transfuse AA patients with red cells (or platelets) from family members if transplantation done from the sibling donor, as this approach may sensitize patients to minor histocompatibility antigens, thereby increasing the risk of graft rejection.
3. Epidemiology
The incidence of AA in the Western countries is approximately 2 per million per year, but it occurs more commonly in the Far East, with a 2 to 3-fold higher[56].A study from France showed that incidence of AAA was approximately 2 per 1,000,000, per year[57]. Another study from Barcelona found that, overall incidence was 2.34 per million inhabitants per year and the incidence increased with age[58]. A Brazilian study indicated that, annual incidence of AA in Parana was 2.4 patients per 1000000 inhabitants[59]. However, in Canada it is 2.4 per million per year among children aged up to 14 years[60]. In China, a study demonstrated that the incidence of AA is approximately 7 per 1,000,000 per year[61]. But another study in China it is projected to be 7.4 patients per 1 million people[62]. The occurrence of AA is 4 per 1,000,000 in Thailand[29]. An epidemiological study found the incidence to be 5 per 1,000,000 in Malaysia[63]. The largest study on the epidemiology was carried out by the International Aplastic Anaemia and Agranulocytosis Study (IAAAS) in Europe and Israel from 1980 to 1984 and found that the overall annual incidence was 2 per million people[62]. The annual incidences of AA in Bangkok and in Thailand were determined to be 4 per million people in the capital and 5.6 per million people in Khonkaen[29]. From various published research article, personal communications, hospital-based studies and first-hand observations it is found that, AA is more prevalent in less developed countries. The geographic variation in its incidence is still unexplained. Only one hospital based study from Lucknow, India, showed the annual incidence of childhood AA was 6.8 per million[64].
4. Potential Mechanisms Responsible For Marrow Failure
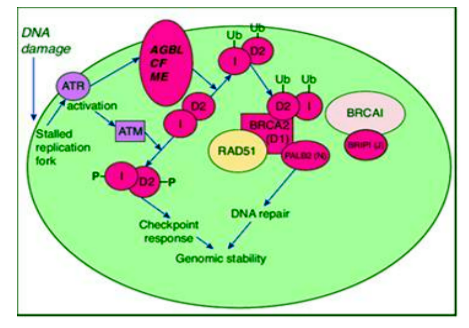
After DNA damage when a replication fork encounters a DNA cross-link, ATR (ataxia Telangiectasia and rad3-related protein) is activated. This leads to the activation of the FA pathway as well as cell-cycle checkpoint activation via the ATM (Ataxia Telangiectasia Mutated) protein. Thirteen complementation groups, defined by somatic cell hybridization, are associated with the development of FA. The complementation groups have been designated as FANC A, B, C, D1, D2, E, F, G, I, J, L, M, and N. The most frequent mutations are found in FANCA, FANCC, or FANCG[65]. It has been proposed that the A and C genes produce cytoplasmic proteins forming a “FA core complex”, whereas the products of genes B, E, F, G, L and M form adaptors or phosphorylators[66,67]. The complex translocates to the nucleus, ubiquitylates FANCD2 (FANCD2-Ub) and FANCI (I-Ub). The I-Ub/FANCD2-Ub complex is then targeted to the chromatin containing the cross link where it interacts with BRCA2 and possibly other DNA repair proteins like BRCA1, FANCD1/BRCA2, FANCN/PALB2, and RAD51[67]. By this FA/BRCA pathway the cells are protected from DNA cross-linking, there by participating in DNA repair mechanism. In the presence of a mutant protein, the normal function is hampered, leading to the damage of hematopoietic stem cells and bone marrow failure.
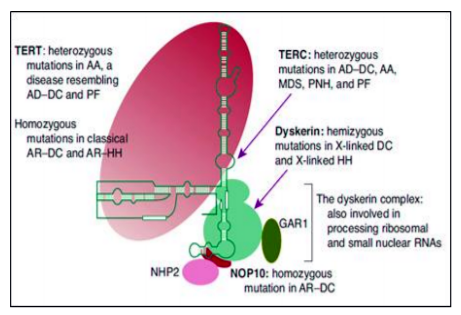
DKC is instigated by telomerase and telomere complex dysfunction. It can be inherited as recessive X- linked disorder but rarely can be autosomal dominant or autosomal recessive. To date, there are 14 genes that have been identified with DKC (ACD, DCK1, TERC, TERT, NOP10, NHP2, TINF2, USB1, TCAB1, CTC1, PARN, RTEL1, WRAP53, and C16orf 57).[68] The telomerase complex which are nucleotide tandem repeat structures (e.g., 5′-TTAGGG-3′) resides at the termini of eukaryotic chromosomes are responsible for maintenance of length of telomeres. Telomerase restores the G-rich telomere repeats which are lost as a result of end-processing during cell division, thus maintain chromosome integrity by preventing end to end fusion, chromosome degradation and chromosome instability. Rapidly proliferating cells are at highest risk for dysfunction and may lead to bone marrow failure. Telomerase is an RNA-protein complex. DKC1gene encodes dyskerin, which is a conserved multifunctional protein component of the telomerase complex.
TERC is the RNA component of the telomerase that is never translated and TERT is the reverse transcriptase. The minimal active telomerase enzyme is composed of two molecules each of TERT, TERC and dyskerin. The TERT uses to synthesize the 6-bp repeats on the 3′ end of telomeric DNA. Dyskerin, GAR1, NHP2, and NOP10 are important proteins for the stability of the telomerase complex. Shelterin complex has six subunits TRF1, TRF2, POT1, RAP1, TIN2, and TPP1 and they can operate in smaller subsets to protect telomere. TINF2 is a component of the shelterin complex, which helps in the distinction of telomeres from sites of DNA damage, preventing their otherwise inappropriate processing[69].
SDS results from mutations in the SBDS gene on chromosome 7q11. It is responsible for cellular apoptosis via the FAS pathway[70]. This
hyper-proliferation results in the abnormal telomere shortening. The pathogenic mechanism that (1) prevents development of pancreatic acinar cells, (2) results in abnormal bone morphogenesis, and (3) causes marrow impairment of blood cell production is still not understood.
Diamond-Blackfan anaemia (DBA) is caused by mutations in the RPL5, RPL11, RPL35A, RPS7, RPS10, RPS17, RPS19, RPS24, and RPS26 genes. These genes provide instructions for making several of the approximately 80 different ribosomal proteins, which are components of cellular structures called ribosomes. Approximately 25 percent of individuals with DBA have identified mutations in the RPS19 gene. Another approximately 25 to 35 percent of individuals with this disorder have been identified with mutations in the RPL5, RPL11, RPL35A, RPS7, RPS10, RPS17, RPS24 or RPS26 genes. In the remaining 40-50% of patients, the cause of the condition is unknown. The specific functions of each ribosomal protein within these subunits are unclear. Some ribosomal proteins are involved in the assembly or stability of ribosomes. Others help in carrying out the ribosome's main function of building new proteins. Studies indicate that a shortage of functioning ribosomal proteins may increase the self-destruction HSC in BM, resulting AA[71].
The common pathway to the clinical disease is a decrease in blood cell formation in the marrow. The number of marrow CD34+ cells (multi-potential hematopoietic progenitors) and their derivative colony-forming unit–granulocyte-macrophage (CFU-GM) and burst forming unit-erythroid (BFU-E) are reduced markedly in patients with AA[72]. A small fraction of patients is initiated by a toxic & drug exposure, or viral infection. But the pathogenesis may also relate to autoimmunity as there is evidence of immune dysfunction in seronegative hepatitis, after benzene exposure. Because many such patients respond to anti–T-cell therapy.The reduced hematopoiesis in most patients of AAA results from the cytotoxic T-cell-mediated immune suppression of very early CD34+ hematopoietic multi-potential progenitor or stem cells. Antigens are presented to T lymphocytes (Th1) by antigen presenting cells (APCs). This triggers T cells activation and proliferation. T-bet, a transcription factor binds to the interferon-γ (IFN-γ) promoter region and induces gene expression. SLAM-associated protein (SAP) binds to Fyn and modulates the signaling lymphocyte activation molecule (SLAM) activity on IFN-γ expression, thus diminishing gene transcription.
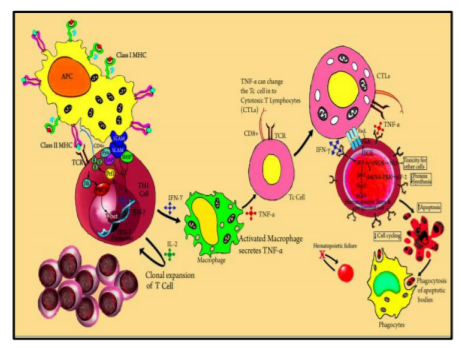
Patients with AA show constitutive T-bet expression with low SAP levels. IFN-γ and tumor necrosis factor-α (TNF-α) up-regulate both the T cell’s cellular receptors and the Fas receptor. Increased production of interleukin-2 leads to polyclonal expansion of T cells. Activation of the Fas receptor initiates apoptosis of target cells by Fas-ligand-Fas-receptor interaction. Some effects of IFN-γ are mediated through interferon regulatory factor 1 (IRF-1), which inhibits the transcription of cellular genes and restricts the cell cycle. IFN-γ is a potent inducer of many cellular genes like inducible nitric oxide synthase (NOS). The production of nitric oxide (NO) may diffuse imparts its cytotoxic effects. These incidences eventually lead to reduced cell cycling and cell death by apoptosis.
5. Manifestations
Common symptoms of this disease are fatigue, shortness of breath, dizziness, and bleeding from the gum, nose, mouth or rectum. People will also bruise easily when this disease is present. A purplish, flat bruise is observed when blood leaks out into the top layers of skin is referred to as an ecchymosis. Patients suffer from pinpoint red spots on their skin and suffer from frequent infections. Maximum, but not all, affected individuals have one or more somatic abnormalities. The absence of typical physical abnormalities like developmental delay, microcephaly, strabismus, abnormality in gastrointestinal system, hypogonadism, café-au-lait spots, absence or malformation of thumb, radius and ulnar bone etc… sometimes makes the clinicians confuse thus the patients remain undiagnosed[73]. Generally FA patients are diagnosed between the age of 3-14 years[74].
Two current studies from the same region focused on the fact that stress cytogenetics is absolutely recommended in AA patients (< 50 years) whether the patient is phenotypically normal or not[75]. Leucoplakia, nail dystrophy and pigmentation of the skin are characteristic of another IAA, which is DKC. The median age was reported 7 years for DKC[76]. Some affected patients may have none of these clinical features and the diagnosis is made later after failure to respond to immunosuppressive therapy[77].
6. Diagnosis And Investigations
According to the guidelines of BCSH and International Agranulocytosis and Aplastic Anaemia Study Group (IAAA) 1987, at least two criteria with hypocellular bone marrow must be present to define AA which are, neutrophil count < 1.5 × 109/L, platelet count < 50×109/L, haemoglobin< 100 g/L, and reticulocyte count < 40×109/L. Depending upon the counts of the cell lineage AA can be classified in to three categories, very severe aplastic anaemia (VSAA), severe aplastic anaemia (SAA), moderately severe aplastic anaemia (MSAA) or non-severe aplastic anaemia (NSAA).
Blood film examination is mandatory to exclude the presence of dysplastic neutrophils and abnormal platelets, blasts cells and hairy cells. Anisopoikilocytosis is common in AA and neutrophils may exhibit toxic granulation. Platelets are reduced in number and size.
HbF should be measured in children before transfusion as this is an important prognostic factor in paediatric MDS. [78]
Often the biopsy of AA shows numerous spicules with empty, fat-filled spaces with relatively few hematopoietic cells. Sometimes spicules are cellular or hypocellular. Hypocellular spicules are called hot spots. Megakaryocytes are reduced. Granulocytic cells generally appear normal. Occasionally mild macro-normoblastic erythropoiesis is found.
AA being a disorder of myloid cell lineage it is important to do the cytogenetics from BM not from PVB. In AA the BM is hypocellular and often insufficient metaphases are obtained. The results of karyotype are generally normal in AA. Clonal cytogenetic abnormalities in otherwise apparent AA are indicative of an underlying hypo-accumulative clonal myeloid disease[79]. It was previously assumed that the presence of an abnormal cytogenetic clone indicated a diagnosis of MDS but not AA, but according to the literatures abnormal cytogenetic clones may be present in up to 12% of patients with otherwise typical AA at diagnosis[79].
Chromosomal breakage test/Stress cytogenetics is performed to identify or exclude FA. PVB lymphocytes should be tested for spontaneous breaks by diepoxybutane (DEB) or mitomycin C (MMC). It is difficult to set an upper age limit for FA screening because it might be diagnosed in the fourth and rarely in the fifth decade, of life[80]. DKC may be excluded by identifying the known gene mutations[81].
Presence of PNH clone must be identified by flow cytometry which is a sensitive and quantitative test for detecting glycosyl- phosphatidylinositol (GPI)-anchored proteins, such as CD55 and CD59[82]. Previously Ham test and sucrose lysis test was used but nowadays flow cytometry is widely performed[83].
It was previously assumed that the presence of an abnormal cytogenetic clone is suggestive of MDS and not of AA, but now various studies have reported that abnormal cytogenetic clones may be present in 4-12% of patients with otherwise typical AA at diagnosis. The presence especially monosomy 7, should alert to the likelihood of MDS. Abnormal cytogenetic clones may also arise during the course of the disease. The most frequently observed abnormalities include trisomy 12, trisomy 8, trisomy 6, and non-numerical chromosomal anomalies i.e. 5q-, 7q-, t (5:12) and 13q-.[84] Often the cytogenetic abnormalities comprise only a small proportion of total metaphases. It may be transient and disappear spontaneously after IST. It has been reported that a good response to IST in patients who acquired a trisomy 8 after treatment compared with a worse prognosis and high risk of leukaemic transformation for patients with monosomy 7[85]. In trisomy 8, the patients respond to immunosuppressive therapies and oligoclonal Tcell expansions are usually present[86] . The anuploid cells specifically express WT1 antigen at high level on their surface. T-cell oligoclones recognize the aneuploidy cells but the target cells are not killed due to up-regulation of antiapoptosis genes, including c-myc, survivin, and CDK1[87].The abnormal trisomy 8 cells,can survive for their capacity to skip the cytotoxic lymphocyte attack. Research findings have recommended that aneuploid clones increase in an abnormal cytokine milieu rich in G-CSF. The presence of a defective G-CSF receptor isoform gives a signal of proliferation, but not differentiation. [88] The presence of even small monosomy 7 clones in the BM, as detected by FISH (but not by routine cytogenetics), is a marker of poor prognosis for response to IST[89]. The literatures have already documented that 4-15% patients with AAA showed cytogenetic abnormalities during diagnosis[90].
Due to differences in diagnostic criteria, patient populations, treatment protocols and the frequency of follow-up BM examinations, acquisition of an abnormal karyotype in AA is frequent, but estimates have been variable between published studies. [91]An early study from Seattle, an abnormal chromosomal profile was observer in 3 of 183 AA patients in the course of the disease. A study from Europe reported, chromosomal abnormality in 23 of 170 patients but in four patients, chromosomal changes were present at the time of diagnosis. In another report of 69 Italian AA patients, an abnormal karyotype was found in 18, but the findings were transient in eight patients and in only seven patients was the karyotypic abnormality arisen during the course of the disease[92]. In a recent series of 13 patients with AA only 2 developed the abnormality later in the course of the disease[93]. A study in japan on 40 children treated with G-CSF and CsA, abnormal cytogenetic evolution was found in 11 patients[94]. Another recent study in USA focused on 122 AA patients treated with IST, among them 14 have developed karyotypic abnormalities with a risk of approximately 21% at 10 years. In a preliminary combined analysis of 189 AA patients treated with various IST regimens on consecutive protocols, the actuarial risk of karyotypic conversion was estimated 14% at 5 years and 20% at 10 years, although it is an unpublished observation[85]. A study from north India showed five (11.9%) paediatric AA patients with chromosomal abnormalities like trisomy 12, trisomy 8, monosomy 7, deletion 7q and translocation (5;12)[95]. A recent study was done from West engal focused on the cytogenetics aspect of AA and emphasized on the fact that cytogenetic study as a routine examination in otherwise typical AA is not essential in this population until the BMA and BMB is showing blasts, dysplasia, a pre-leukemic changes, fibrosis etc...[96]
To reject the possibility of megaloblastic anaemia, Vitamin B12 and folate levels should be measured before a final diagnosis of AA.
Liver function tests (LFT) must be performed to detect antecedent hepatitis.
In most post-hepatitic AA the serology is most often negative for all the known hepatitis viruses (Non-A, - B, -C, -D, -E, or -G hepatitis virus). The onset of the disease occurs after a period of 2–3 months of acute hepatitis and is more common in young males[97]. Tests should be performed for hepatitis A antibody, hepatitis B surface antigen, hepatitis C antibody and Epstein–Barr virus (EBV). CMV and HIV should be evaluated if BMT is planned.
Blood should be tested for anti-nuclear antibody and anti-DNA antibody in AA patients.
Chest X-ray is useful to exclude infection. X-ray of the radii indicates the presence of FA but chromosomal breakage test must be performed to diagnose FA. The enlarged spleen and/or enlarged lymph nodes in Ultrasonography may indicate the probability of malignant haematological disorder. Abnormal or anatomically displaced kidneys are features of FA.
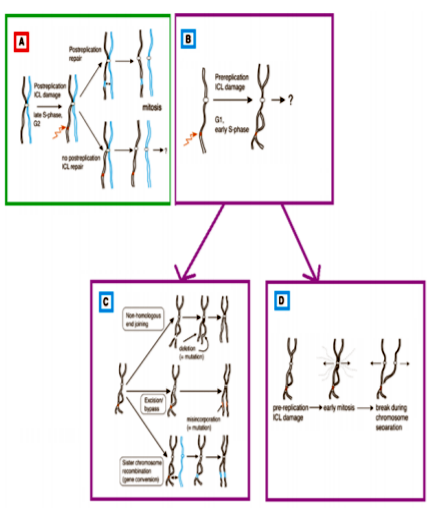
A chromosome breakage test or Stress Cytogenetics with mitomycin-c or diepoxybutane is done in AA patients who are < 50 years old to exclude FA[98]. The most consistent and accepted cellular phenotype of FA patients, is hypersensitivity to DNA crosslinking agents like mitomycin C (MMC) or diepoxybutane (DEB) which produce Interstrand crosslinks (ICLs)[99]. Chromosome breakage and radial formation are nothing but Interstrand crosslinks (ICLs). It can occur before and after DNA replication in the homologous region of sister chromatid. If ICLs occur in post-replication state, two possibilities are there. Either homologous repair between sister chromatids produce an error free repair or mitosis proceeds normally and one of the daughter cells inherits an unrepaired ICL. In case ICLs occur in the pre replication state, a homologous sister chromatid is not available for re-combinational repair. There are three possible mechanisms for repair of pre-replication ICLs[100] i) Non-homologous end joining, ii) Excision repair and/or lesion bypass and iii) Homologous repair between homologous chromosomes. Whereas first two mechanisms are error-prone, the third one is error-free. Unrepaired ICLs generate a mandatory chromatid-break during mitosis[100,101].
Another important aspect of this disease is the cytokines. Cytokine plays a significant role in the process of BM failure. Cytotoxic T-cell- mediated attack on multi potential hematopoietic stem cells (HSC) is the basis for AAA. Immune injury to the marrow occurs after exposure to etiological agents which react as neo-antigens that provoke a secondary T-cell-mediated attack of HSC. Elevated serum levels of IFN-γ are present in approximately 30 percent of patients with AA and IFN-γ expression has been detected in the marrow of most AAA patients[102]. Addition of antibodies to IFN-γ enhances in vitro colony growth of marrow cells in affected patients. These observations indicate that AAA is the result of cellular immune-induced apoptosis of primitive CD34+multi-potentialhematopoietic progenitors, mediated by cytotoxic T lymphocytes, in part, through the expression of T-helper type1 (Th1) inhibitory cytokines [103] like IL-2 [104], IFN-γ[104]and TNF-α [105] Current evidence indicates that auto reactive T cells, activated by different stimuli, release myelo-suppressive cytokines including TNF- α and IFN- γ which cause HSC death by blocking mitosis and increasing apoptosis by Fas ligand-Fas receptor interaction[106]. That is why these cytokines also defined as anti-hematopoietic cytokines. A study was done in 2010 in USA on the signature profiles of cytokine in AAA and MDS. This study emphasized on the signature profile of cytokines in AA, which showed high levels of thrombopoietin and granulocyte colony-stimulating factor, with low levels of CD40 ligand, chemokine (C-X-C motif) ligand 5, chemokine (C-C motif) ligand 5, chemokine (C-X-C motif) ligand 11, epidermal growth factor, vascular endothelial growth factor and chemokine (C-C motif) ligand 11. High levels of TNF-α, interleukin-6, chemokine (C-C motif) ligand 3, interleukin-1 receptor antagonist, and hepatocyte growth factor were a cytokine signature for MDS[107]. Another research focused on the fact that decreased plasma cytokines are associated with low platelet counts in AA and immune thrombocytopenic purpura[108]. A different kind of study focused on the successful cytokine treatment of aplastic anaemia following living-related orthotopic liver transplantation for non-A, non-B, non-C hepatitis[109]. Another study with Lymphocytic Choriomeningitis Virus showed exhaustion of cytokine-secreting CD8+ T Cells in persistent infection in AA patients[110]. A Japanese study found that the levels of IL-6, IL-1β and TNF-α in cultured peripheral blood mononuclear cells were higher in AA or in refractory anaemia (RA) than in RA with excess of blasts (RAEB) or in RAEB in transformation (RAEB-T).
Marked cytokine overproduction was observed in RA as well as in AA (Koike et al. 1995)[111]. Some study have emphasized on the molecular aspect of cytokine genes and found that IFN-γ +874 A/T gene polymorphism is associated with three-fold increased risk of development of AA[112].
Few Indian studies are present depicting the profile of cytokine expression of IL-8, IFN-γ and TNF-α in BM T- cells and their levels in bone marrow plasma in patients with AA[113]. Studies have concluded that increased production of both IFN-γ and TNF-α in the BM may induce disease pathogenesis in AA and ATG therapy which may contribute to hematological remission by decreasing the elevated levels of IFN-γ and TNF-α in BM.Recently a comparative study of bone marrow and blood plasma levels of IL-2 in AA was done and revealed that it is associated with the disease severity[114].
Previously nonrandom X-chromosome inactivation (NRXI) or cytogenetics was used to detect the abnormal clone in AA. Now next-generation DNA sequencing (NGS), together with the existing knowledge can give us a constructive opinion about the major causative mutations in MDS/AML and AA, further relating AA to the clonal evolution of MDS/AML.There are neumerous NGS-based mutation studies on AA, reported varying mutation frequencies ranging from 5.3% to 72%[115]. A comprehensively mutation study using targeted-capture deep sequencing was performed on 106 genes commonly mutated in myeloid cancers[116]. The average allelic burden of mutations was considerably higher in MDS (75%) than AA (10%). No evidence of lower allele frequency was examined after IST as compared to the time of diagnosis. Mutation analysis for the known genes FANCA,FANCC, FANCDI (BRCA2,) FANCD2, FANCE, FANCF, FANCG (XRCC9), FANCI (KIAA1794), FANCJ (BACH1/BRIP1), FANCL (PHF9/ POG), FANCM (Hef), FANCN (PALB2),TERC & TERT should be done to exclude FA and DKC.[81]Telomere lengths can be measured although not available as a routine clinical service. Mutations are usually present in multi-lineage compartments, including HSCs, common myeloid progenitors and erythroid progenitors, in a minor fraction of T cells, proposing that the mutations occurred at a stem cell level. PIGA mutations, is a somatic mutations were detected 19.3% patients[117]. Mutations are more commonly observed in older patients and those suffering from longer than 6 months.
Having a pancytopenia does not always make the decision of AA. It must be critically judge before the starting the treatment regimen. There are other condition like pancytopenia with hypocellular bone marrow other than AA (MDS, AML, ALL etc…), pancytopenia with cellular bone marrow (Primary bone marrow diseases, Myelodysplasia syndromes, PNH, myelofibrosis, some aleukemic leukemias, myelophthisis, bone marrow lymphoma etc…) and hypocellular bone marrow ± cytopenia (Q fever, Legionnaires disease etc…)
7. Treatment
Red cell and platelet transfusions are essential in AA patients to maintain a safe blood count. It is advised to give prophylactic platelet transfusions when the platelet count is < 10 ×109/l (or < 20 × 109/l in the fever), rather than giving platelets alone in presence of bleeding manifestations[118]. Red cell transfusions should be given to maintain a safe haemoglobin level >80 g/l. In multi-transfused patients with AA there is an obvious chance of allo-immunisation to leucocytes present in red cell and platelet transfusions by generating HLA or non- HLA (minor histocompatibility) antibodies. This can create platelet refractoriness, as well as an increased risk of graft rejection after allogeneic BMT[119]. Pre-storage leuco-depleted units of red cells and platelets can reduce the risk of allo-immunisation[120]. Patients who become refractory to platelet transfusions should be screened for HLA antibodies. AA patients can become sensitised to random donor platelets resulting in platelet refractoriness, in such a situation HLA-matched platelets should be used. Blood product from the family members are restricted because the recipient may become sensitised to minor histocompatibility antigens from the potential bone marrow donor resulting increased chance of graft rejection. In an exceptional circumstance, like urgent platelet transfusion, the patient may develope multi-specific HLA antibodies.
The patient who is a BMT candidate must be transfused with CMV-negative blood products until the patient’s CMV status is known. Then CMV-negative blood products can be continued only if both the patient and donor is CMV negative[121].
Granulocyte transfusions can be used in neutropenia. Adverse effects, such as febrile reactions, HLA allo-immunization and transfusion-related acute lung injury (TRALI) are reported complications after granulocyte transfusions. The use of irradiated granulocyte transfusions should be restricted in the patients, if it becomes hazardous.
Till now no effective and safe haemopoietic growth factor has been reported which can support red cell and platelet counts in AA patients[122]. Use of rHuEpo in AA is ineffective[123]. Interleukin-6 (IL-6) has been evaluated as a factor evolving severe anaemia and haemorrhage[124]. Stem cell factor was shown to stimulate tri-lineage haemopoiesis in some patients with AA[125]. Use of stem cell factor with ATG and cyclosporin is debatable because of serious toxicity from anaphylaxis reactions. The developments of anti-TPO antibodies and prolonged thrombocytopenia have been reported against the truncated version of rHu-TPO, supports discontinuation of its use in clinical trials[126]. Second generation thrombopoiesis stimulating agents have not undergone clinical trials in AA. No controlled studies have been reported that G-CSF or other haemopoietic growth factors are helpful in the treatment of severe infection in patients with AA. A short course of subcutaneous G-CSF at a dose of 5 lg/ kg per day may be considered for severe systemic infections when response to intravenous antibiotics and antifungals drugs are absent. GM-CSF is not generally recommended as it can induce severe haemorrhage and other serious toxicity.
The risk of infection is determined by the neutrophil and monocyte counts[127]. Patients with aplastic anaemia are at risk of bacterial and fungal infections[128]. Initially a synergistic combination of antibiotics, such as an aminoglycoside and a,b-lactam penicillin, is the exact choice depending on local hospital microbiological sensitivity/resistance patterns. The duration of neutropenia, infection history and current antibiotics help in the choice of antibiotic, including the early introduction of amphotericin. Pulmonary infiltrates and sinus infection are the indicators of likely fungal infection in patients with SAA. Aspergillus infection is very common in patients with SAA because of the frequent prolonged periods of severe neutropenia and monocytopenia.
Severely neutropenic (< 0.5 ×109/l) patients should preferably be nursed in isolation when in hospital and should receive prophylactic antibiotics and antifungals. A regular antiseptic mouthwash, such as chlorhexidine must be used with a low bacterial content diet[128]. Prophylactic antibiotics neomycin, colistin, quinolone antibiotic, such as ciprofloxacin are given to help prevent Gram-negative sepsis. Prophylactically ciprofloxacin cannot be used in febrile neutropenic episodes. In persistent fever systemic antifungal therapy is introduced earlier into the febrile neutropenia regimen. However, nowadaysappearance of quinoloneresistant bacteria, increase in Gram-positive infections arecontributing to the infections. Itraconazole and posaconazole are the choice of drugs to treat Aspergillus infection, whereas Fluconazole does not provide the coverage.
Iron overload can cause major problems in heavily transfused patients. Subcutaneous desferrioxamine should be started when the serum ferritin is >1000 lg/l, although there is very little evidence[129]. Instead of subcutaneous intravenous desferrioxamine may be considered if subcutaneous desferrioxamine is not tolerated. The routine use of oral iron chelator deferiprone is not recommended in patients with AA AS it may cater Agranulocytosis[129].
Some sketchy rumour of vaccination producing BM failure or triggering relapse of AA has been reported. All live vaccines should be avoided after BMT and ATG treatment. Vaccinations should only be given in absolutely necessity[130].
The standard specific treatment for is used in AA patients, either allogeneic stem cell transplantation from an HLA-identical sibling donor or immunosuppressive therapy with a combination of ATG and cyclosporine. Bleeding and infection should be controlled before specific treatment is given to the patient. Immunosuppressive therapy must not be given in the presence of infection or uncontrolled bleeding. The outcome of stem cell transplantation can also be hampered by infection.
It is suggested that BM stem cells should be collected from BM but not G-CSF mobilized peripheral blood stem cells (PBSC)[131]. It is asier to obtained BM than PBSC and also BM is devoid of the exposure of G-CSF[132]. The standard dose is 3 ×108 nucleated marrow cells/kg because at lower doses the risk of graft rejection increases significantly[133]. A retrospective study from the EBMT of patients undergoing HLA-identical sibling or HLA-identical unrelated donor BMT for aplastic anaemia showed significantly better response in the patients with donors from the same sex. In a small number of AA patients with umbilical cord blood as an alternative source of stem cells for transplantation, although it is associated with a lower risk of acute and chronic GVHD[134].
Umbilical cord blood transplantation can also be considered in pediatric patients who do not have an HLA-identical sibling donor or a fully matched unrelated adult donor.
The initial treatment of choice for newly diagnosed AA patients is allogeneic BMT from an HLA-identical sibling donor.
i. The patient has SAA or VSAA
ii. Patient’s age is < 40 years and has an HLA compatible sibling donor
iii. For children who have NSAA and in whom treatment is indicated
iv. BMT may be considered for patients>40 years who have unsuccessful immunosuppressive therapy and in good medical condition, with HLA compatible donor.
v. There are currently insufficient data available on the use with HSCT in SAA patients >60 years of age[135].
The use of high-resolution, HLA typing of a matched, unrelated donor markedly improves the prognosis for transplantation[136]. High-resolution DNA matching at HLA-A, -B, -C, and -DRB1 (8 of 8 allele) is considered the lowest level of matching consistent with the highest level of survival. If there is an HLA mismatch at one or more loci, especially HLA-A or -DRB1, the outcome is compromised, and immunosuppression with combined therapy may be preferred initially, depending on patient age, cytomegalovirus status and disease severity. There is an argument about the upper age limit for BMT. Outcomes of BMT using an HLA identical sibling donor are poorer in patients >30 years than patients < 30 years of age[137]. BM Transplantation from an HLA identical sibling donor has a chance of long term cure in 75–90% patients[139]. By adequate dose of cyclophosphamide with ATG conditioning, graft failure is approximately 4–14%. Acute GVHD is 12–30 % whereas chronic GVHD occurs in 30–40% of patients. Treatment with immunosuppressive therapy prior to BM transplantation is associated with a poor outcome and increased graft rejection[138].
The conditioning regimen and GVHD prophylaxis are different in AAA and IAA[139]
i.Conditioning regimen for patients aged < 30 years
High dose cyclophosphamide 50 mg/kg×4 (day -5 to -2) and ATG (Thymoglobuline, Genzyme 1.5 vials/10 kg × 3 on days -5 to - 3), with methylprednisolone (not in pediatric patient) 2 mg/kg × 3 (day -5 to -3) is the standard regimen. The recommended post-transplant immunosuppression is (a) cyclosporin 5 mg/kg per day, in two divided doses (that is, 2.5 mg/kg BD), starting on day+1, and continuing for 12 months with tapering beginning at 9 months to help prevent late graft failure, (b) short course methotrexate 15 mg/m2 on day +1, then 10 mg/m2 on days +3, +6, and +11[140].
ii.Conditioning regimens for patients aged >30 years
The patients (age 30 - 50 years), who are potential transplant candidates; the best conditioning regimen is unknown. Patients who are >40 years of age and who are medically suitable for BMT, may receive a reduced intensity conditioning regimen, using cyclophosphamide 1200 mg/m2, fludarabine 120 mg/m2 and either ATG or Alemtuzumab[141].
Matched unrelated donor BMT (MUD BMT) is a new approach in the treatment of SAA. MUD BMT may be considered when patients fulfill all the following criterias.
i. Patients must have a fully matched (at DNA level for both class I and II antigens) donor
ii. Patients’ age must be < 50 years (although if the patient is in good performance status, 50–60 years may be considered)
iii. Patients have at least one course of unsuccessful ATG and cyclosporin (for both adults and children)
iv. In adults a second course of ATG may be favored before proceed to MUD BMT. This is depending on individual patient circumstances for an example patient is having SAA or VSAA with no evidence of infection and/or acute bleeding at time of BMT
v. It is used as first line treatment in IAA patients who does not have an HLA matched sibling donor
There is a significant risk of late GVHD in AA patients after allogeneic BMT. In most of the patients, it is associated with discontinuing cyclosporin too early or low dose of cyclosporin blood levels. Progressive mixed chimaerism indicates towards a high risk of whereas stable mixed chimaerism is associated with excellent survival and a low risk of the same[142].
Chimaerism must be monitored principally during the time of cyclosporin withdrawal. If there is evidence of significant mixed chimaerism (>10% of recipient cells) or an increasing proportion of recipient cells (>15% increase over 3 months), indicate towards a high risk of late rejection and cyclosporin should not be reduced or withdrawn at that time[142]. Chimaerism can be assessed from the short tandem repeats in the mononuclear cells with the sensitive techniques such as polymerase chain reaction (PCR) analysis[143].
Antilymphocyte Serum and Antithymocyte Globulin ATG and ALG act principally by reducing cytotoxic T cells. This involves ATG-induced apoptosis through both FAS and TNF pathways[144]. Cathepsin B also plays a role in T-cell cytotoxicity at clinical concentrations of ATG but may involve an independent apoptosis pathway[145]. ATG and ALG also release hematopoietic growth factors from T cells[146]. Horse and rabbit ATG are licensed in the United States. Skin tests against horse serum should be performed prior to administration[147].
Antithymocyte globulin (ATG) is an immunosuppressive drug and it is used in severely neutropenic patients. The standard preparation of ATG was horse ATG (Lymphoglobuline; Genzyme). The rabbit preparation (Thymoglobuline; Genzyme) (Lymphoglobuline) was withdrawn due to complications in manufacturing. Rabbit ATG (Thymoglobuline) is therefore the first choice of treatment. Response rates to rabbit ATG are expected to be comparable to horse ATG, because both preparations have the same immunogen (thymocytes), similar production method and share similar epitopes for binding. The eligibility criteria are as follows;
i. NSAA patients who are dependent on red cell and/or platelet transfusions
ii. NSAA patients who, although are not transfusion-dependent but may have significant neutropenia and be at risk of infection
iii. SAA or VSAA patients who are >40 years of age and younger SAA or VSAA patients who lack an HLA-compatible sibling donor
IST using the combination of ATG and cyclosporin is associated with response rates of between 60% - 80% with current 5 year survival rates of approximately 75–85%. For SAA,the survival and response rate to ATG alone is significantly less than with the combination of ATG and cyclosporin. In NSAA patients the response to the combination of ATG and cyclosporin is significantly greater than with cyclosporin alone[148].
The response and survival rates are low in older compared with younger patients. The response rate for patients aged >60, 50–59 and < 50 years is 37%, 49% and 57%, and 5-year survival is 50%, 57% and 72%, respectively[149]. For patients aged >70 years, the 10-year survival rate is 33% and patients aged 50 -70 years, the 10-year survival rate is 60%. Patients older than 60 years also have a higher risk of serious cardiac events after ATG[150].
Often insignificant abnormal cytogenetic clone (ACC) may be observed in the AA patients. It comprises only a minor proportion of entire metaphases. It may be transient and disappear spontaneously or after haematological response to successful IST.[83] Patients with ACC should be treated with IST (ATG and cyclosporin) and must not receive chemotherapy as it may result predictable deteriorating of pancytopenia with the probability of irreversible marrow failure. If the patient accomplishes the criteria for sibling BMT, then they should be transplanted. It has been reported that, the response rates to IST appear to be similar to AA patients with or without an ACC. [92]AA patients (who are not BMT candidates) with or without ACC should be managed in the same way (except monosomy 7). A myeloablative regimen may be preferable in patients who are having monosomy 7. A good response to IST has been observed in patients who acquired a trisomy 8[85]after treatment compared with a worse prognosis and high risk of leukaemic transformation for patients with monosomy 7. For other chromosome abnormalities (expect monosomy 7), there is no such documentation which supports myeloablative regimen approach, as the disease appears to follow a course similar to that of AA without ACC[85].
Patients with AA may later develop haemolytic PNH and conversely patients with haemolytic PNH can later progress to AA[83]. Regular folic acid supplementation 5 mg/day must be obtained. Oral iron supplementation should be given to the patients of intravascular haemolysis, but cautiously, as it can trigger acute haemolysis. Prednisolone helps to reduce the degree of haemolysis but an alternate day regimen at low dose of between 10 and 15 mg is used as there are many side effects. An oral cyclosporin which is a complement C5 blocking monoclonal antibody, has been found to be effective at reducing haemolytic PNH[151]. It is still reasonable to consider ATG treatment for patients with a significant PNH clone (>50%). HLA-identical sibling BMT for haemolytic PNH is only prescribed in those patients who later develop SAA or patients with multiple venous thromboses.[152]The effectiveness of Eculizumab reduces venous thromboses but also remove the indication for BMT[153]. The decision of using anticoagulation in AA patients with a significant PNH clone (>50% in granulocytes)[154] is still controversial.
There is a 33% of relapse of AA in pregnancy. Again there is a significant risk of relapse of the disease in pregnancy who has earlier responded to immunosuppressive[155]. Literatures said that after successful allogeneic BMT, pregnancy does not cater relapse of the disease[156]. In the pregnancy period Hb levels and platelet counts decreased while neutrophil counts increased. However, it is important to discuss with the patient and family the potentially serious risks to both the mother and baby. Supportive care is the treatment option for AA patients in pregnancy and the platelet count should be maintained above 20×109/l[157]. Red cell and platelet transfusions during normal pregnancy increase risk of alloimmunisation. ATG is too hazardous to treat AA during pregnancy[46].
8. Conclusion And Future Prospects
Allogeneic stem-cell transplantation or immunosuppression, whatever is the procedure, treatment of AA has developed intensely over the last two and half decades. That enhances the chance of long-term survival of the patients and now in more than 75% patients, the rate of survival can be predicted more accurately with either therapy. For transplantation, extension of stem-cell replacement is the primary challenge for all the patients, regardless of their age, availability of a histocompatible sibling, and to others who don’t have any family alternative of stem-cell sources. The possibility to attain engraftment under these circumstances may involve extensive conditioning regimens in which worries, particularly of any further malignancies, can be averted for a considerable period of time. Moreover, donor selection basing upon high-resolution histocompatibility typing can result more optimistic and favorable results. The successful attainment of umbilical cord-blood transplantations, with a lower possibility of GVHD, may be improved by greater pools and histocompatibility matching. For immunosuppression, a significant number of new medications and treatmentsis in the queue for being tested in AA patients. But, the escalation of expenditures has to be sensibly balanced by the authority. Recurrent use of horse ATG followed by rabbit ATG, alemtuzumab or cyclophosphamide as immunosuppressive therapy; provide the opportunity of blood-count restoration in more than 90% of AA patients. If remaining stem-cell numbers are restraining, ex vivo expansion of haematopoiesis using Hox box proteins can be tried. Quantitative and qualitative assays of oligoclonal T-cell and hematopoietic stem-cell function can improve the judgments of the treatment procedure. Studies on the microenvironment of the marrow can be helpful for the upflifment of the existing knowledge.
Especially the molecular study of antihematopoitic cytokines must be focused. Nowadays study on the genetic, environmental, lifestyle risk factors is important to clarify how these agents can initiate and propagate the marrow destruction in AA. Further researches are needed both for academic and professional purpose in individual and collective level.
9. Acknowledgments
We are thankful to Swami Nityakamananda, Secretary, Ramakrishna Mission Seva Pratishthan, Vivekananda Institute of Medical Sciences, Kolkata and Department of Science of Technology-West Bengal. We are also obliged to our colleagues from the Department of Hematology, Nil Ratan Sircar Medical College and Hospital, Kolkata. The authors are apologized, whose papers were not cited in the bibliography due to space constraints.
References
- Marsh JCW, Ball SE, Cavenagh J, Darbyshire P, Dokal I, Gordon-Smith EC, Keidan J, Laurie A, Martin A, Mercieca J, Killick SB, Stewart R, Yin JAL. Guidelines for the diagnosis and management of aplastic anemia.British Journal of Haematology.2009; 147:43-70.
- Rai A, Vaishali V, Naikmasur VG, Kumar A, Sattur A. Aplastic anemia presenting as bleeding of gingiva: Case report and dental considerations. The Saudi Journal for Dental Research.2016; 7: 69–72.
- Young NS, Maciejewski JP. Williams Hematology: Basic Principles and Practice. 6th ed. Elsevier Saunders, Philadelphia: 2012, ch 28,463-482.
- Fanconi G. Familiare, infantile perniciosähnliche anämie (perniziöses blutbild undkonstitution)[Familialinfantile pernicious-like anemia (pernicious blood picture and constitution)]. Jahrbuch für Kinderheilkunde und Physische Erziehung.1927;117:257-280.
- Sagaseta de Ilurdoz M, Molina J, Lezáun I, Valiente A, Durán G. Updating Fanconi's anaemia. An Sist Sanit Navar.2003; 26 (1):63-78.
- Rosendorff J, Bernstein R, Macdougall L, Jenkins T. Fanconi anemia: another disease of unusually high prevalence in the Afrikaans population of South Africa. Am J Med Genet.1987; 27(4):793-797.
- Dokal I, Vulliamy T. Inherited aplastic anemias/bone marrow failure syndromes. Blood Reviews.2008;22(3):141–153.
- Shwachman H, Diamond LK, Oski FA, Khaw KT. The syndrome of pancreatic insufficiency and bone marrow dysfunction.J Pediatr. 1964;65:645-63.
- Aggett PJ, Cavanagh NP, Matthew DJ, Pincott JR, Sutcliffe J, Harries JT. Shwachman's syndrome. A review of 21 cases. Arch Dis Child.1980;55(5):331-347.
- Kaushansky K, Lichtman M, Beutler E, Kipps T, Prchal J, Seligsohn U. Williams Hematology: Basic Principles and Practice.6th ed. McGraw-Hill Medical, New York: 2010 ch 35.
- Diamond LK, Blackfan KD. Hypoplastic anemia. Am. J. Dis. Child. 1938;56:464–467.
- Vlachos A, Ball S, Dahl N, Alter BP, Sheth S, Ramenghi U, Meerpohl J, Karlsson S, Liu JM, Leblanc T, Paley C, Kang EM, Leder EJ, Atsidaftos E, Shimamura A, Bessler M, Glader B, Lipton JM; Participants of Sixth Annual Daniella Maria Arturi International Consensus Conference.Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol. 2008;142(6):859-876.
- Li FP, Hecht F, Kaiser-McCaw B, Baranko PV, Potter NU. Ataxia-pancytopenia: syndrome of cerebellar ataxia, hypoplastic anemia, monosomy 7, and acute myelogenous leukemia. Cancer Genet Cytogenet.1981;4(3):189-196.
- Geddis AE. Inherited thrombocytopenia: Congenital amegakaryocytic thrombocytopenia and thrombocytopenia with absent radii. Semin Hematol. 2006;43(3):196-203.
- Pierce AJ, Jasin PM. NHEJ deficiency and disease. Mol Cell.2001;8(6):1160-1161.
- Walters TR, Desposito F. Aplastic anemia in Dubowitz syndrome. J Pediatr. 1985;106(4):622-623.
- Gennery AR, Slatter MA, Bhattacharya A, Barge D, Haigh S, O'Driscoll M, Coleman R, Abinun M, Flood TJ, Cant AJ, Jeggo PA. The clinical and biological overlap between Nijmegen Breakage Syndrome and Fanconi anemia. Clin Immunol. 2004;113(2):214-219.
- Stephan JL, Vlekova V, Le Deist F, Blanche S, Donadieu J, De Saint-Basile G, Durandy A, Griscelli C, Fischer A. Severe combined immunodeficiency: a retrospective single-center study of clinical presentation and outcome in 117 patients. J Pediatr.1993;123(4):564-572.
- Espérou-Bourdeau H, Leblanc T, Schaison G, Gluckman E. Aplastic anemia associated with ''bird-headed'' dwarfism (Seckel syndrome). Nouv Rev Fr Hematol.1993;35:99-100.
- Gonzalez CH, Durkin-Stamm MV, Geimer NF, Shahidi NT, Schilling RF, Rubira F, Opitz JM. The WT syndrome--a "new" autosomal dominant pleiotropic trait of radial/ulnar hypoplasia with high risk of bone marrow failure and/or leukemia. Birth Defects Orig Artic Ser. 1977;13(3B):31-38.
- Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108(8):2509-2519.
- Snyder R. Benzene's toxicity: a consolidated short review of human and animal studies by HA Khan. Hum Exp Toxicol. 2007;26(9):687-696.
- Rugman FP, Cosstick R. Aplastic anaemia associated with organochlorine pesticide: case reports and review of evidence. J Clin Pathol. 1990;43(2):98-101.
- Prager D, Peters C: Development of aplastic anemia and the exposure to Stoddard solvent. Blood. 1970;35(3):286-7.
- Sabbioni G, Sepai O, Norppa H, Yan H, Hirvonen A, Zheng Y, Järventaus H, Bäck B, Brooks LR, Warren SH, Demarini DM, Liu YY. Comparison of biomarkers in workers exposed to 2,4,6-trinitrotoluene. Biomarkers. 2007;12(1):21-37.
- Crawford M.A.D: Aplastic Anaemia Due to Trinitrotoluene Intoxication. Br Med J. 1954; 2(4885): 430–437.
- Powars D. Aplastic anemia secondary to glue sniffing. N Engl J Med. 1965;273(13):700-702.
- Muir KR, Chilvers CE, Harriss C, Coulson L, Grainge M, Darbyshire P, Geary C, Hows J, Marsh J, Rutherford T, Taylor M, Gordon-Smith EC. The role of occupational and environmental exposures in the aetiology of acquired severe aplastic anaemia: a case control investigation. Br J Haematol. 2003;123(5):906-14.
- Issaragrisil S, Kaufman DW, Anderson T, Chansung K, Leaverton PE, Shapiro S, Young NS. The epidemiology of aplastic anemia in Thailand.Blood. 2006;107(4):1299-307.
- Ajlouni K, Doeblin TD: The syndrome of hepatitis and aplastic anaemia. Br J Haematol.1974; 27(2): 345-355
- Pol S, Driss F, Devergie A, Brechot C, Berthelot P, Gluckman E. Is hepatitis C virus involved in hepatitis-associated aplastic anemia? Ann Intern Med. 1990;113(6):435-437.
- Mishra B, Malhotra P, Ratho RK, Singh MP, Varma S, Varma N. Human parvovirus B19 in patients with aplastic anemia. Am J Hematol. 2005;79(2):166-167.
- Wong S, Young NS, Brown KE. Prevalence of parvovirus B19 in liver tissue: no association with fulminant hepatitis or hepatitis-associated aplastic anemia. J Infect Dis. 2003;187(10):1581-1586.
- Lazarus KH, Baehner RL. Aplastic anemia complicating infectious mononucleosis: a case report and review of the literature. Pediatrics. 1981;67(6):907-910.
- Vinters HV, Mah V, Mohrmann R, Wiley CA. Evidence for human immunodeficiency virus (HIV) infection of the brain in a patient with aplastic anemia. Acta Neuropathol. 1988;76(3):321-4.
- Rosenfeld CS, Rybka WB, Weinbaum D, Carrigan DR, Knox KK, Andrews DF, Shadduck RK. Late graft failure due to dual bone marrow infection with variants A and B of human herpesvirus-6. Exp Hematol. 1995;23(7):626-629.
- BrodskyRA.Paroxysmalnocturnal hemoglobinuria.Blood. 2014;124(18):2804-2811.
- Pavithran K, Raji NL, Thomas M. Aplastic anemia complicating systemic lupus erythematosus--report of a case and review of the literature.Rheumatol Int. 2002;22(6):253-255.
- Bailey FA, Lilly M, Bertoli LF, Ball GV. An antibody that inhibits in vitro bone marrow proliferation in a patient with systemic lupus erythematosus and aplastic anemia.Arthritis Rheum. 1989;32(7):901-905.
- Dinçol G, Saka B, Aktan M, Nalçaci M, Keskin H, Palanduz S, Oztürk S, Dinçol K. Very severe aplastic anemia following resection of lymphocytic thymoma: effectiveness of antilymphocyte globulin, cyclosporin A, and granulocyte-colony stimulating factor. Am J Hematol. 2000;64(1):78-9.
- Roffe C, Cahill MR, Samanta A, Bricknell S, Durrant ST. Aplastic anaemia in systemic lupus erythematosus: a cellular immune mechanism? Br J Rheumatol. 1991;30(4):301-304.
- Kim SW, Rice L, Champlin R, Udden MM. Aplastic anemia in eosinophilic fasciitis: responses to immunosuppression and marrow transplantation. Haematologia (Budap). 1997;28(3):131-137.
- Kumar M, Goldman J. Severe aplastic anaemia and Grave's disease in a paediatric patient. Br J Haematol. 2002 Jul;118(1):327-329.
- Abrams EM, Gibson IW, Blydt-Hansen TD. The concurrent presentation of minimal change nephrotic syndrome and aplastic anemia. Pediatr Nephrol. 2009;24(2):407-409.
- Ehrlich P. Über einen Fall von Anamie mit Bemerkungen über regenerative Veranderungen des Knochenmarks. Charite Ann.1888;300-309.
- Aitchison RG, Marsh JC, Hows JM, Russell NH, Gordon-Smith EC. Pregnancy associated aplastic anaemia: a report of five cases and review of current management. Br J Haematol. 1989;73(4):541-545.
- Tichelli A, Socié G, Marsh J, Barge R, Frickhofen N, McCann S, Bacigalupo A, Hows J, Marin P, Nachbaur D, Symeonidis A, Passweg J, Schrezenmeier H; European Group for Blood and Marrow Transplantation Severe Aplastic Anaemia Working Party. Outcome of pregnancy and disease course among women with aplastic anemia treated with immunosuppression. Ann Intern Med. 2002;137(3):164-72.
- Nimer SD, Leung DH, Wolin MJ, Golde DW. Serum stem cell factor levels in patients with aplastic anemia. Int J Hematol. 1994;60(3):185-189.
- Lyman SD, Seaberg M, Hanna R, Zappone J, Brasel K, Abkowitz JL, Prchal JT, Schultz JC, Shahidi NT. Plasma/serum levels of flt3 ligand are low in normal individuals and highly elevated in patients with Fanconi anemia and acquired aplastic anemia. Blood. 1995;86(11):4091-96.
- Kojima S, Matsuyama T, Kodera Y, Nishihira H, Ueda K, Shimbo T, Nakahata T. Measurement of endogenous plasma granulocyte colony-stimulating factor in patients with acquired aplastic anemia by a sensitive chemiluminescent immunoassay. Blood. 1996;87(4):1303-8.
- Kojima S, Matsuyama T, Kodera Y. Circulating erythropoietin in patients with acquired aplastic anaemia. Acta Haematol. 1995;94(3):117-22.
- Emmons RV1, Reid DM, Cohen RL, Meng G, Young NS, Dunbar CE, Shulman NR.Human thrombopoietin levels are high when thrombocytopenia is due to megakaryocyte deficiency and low when due to increased platelet destruction. Blood. 1996;87(10):4068-4071.
- Nakao S1, Matsushima K, Young N. Decreased interleukin 1 production in aplastic anaemia. Br J Haematol. 1989;71(3):431-436
- Holmberg LA, Seidel K, Leisenring W, TorokStorb B. Aplastic anemia: analysis of stromal cell function in long-term marrow cultures. Blood. 1994;84(11):3685-90.
- Sutton JF, Stacey M, Kearns WG, Rieg TS, Young NS, Liu JM. Increased risk for aplastic anemia and Myelodysplastic syndrome in individuals lacking GSTT1 gene. Pediatric Blood Cancer. 2004;42:122-126.
- Ehsan A, Shah SA, Ibrahim T. Epidemiology of acquired aplastic anaemia in Pakistan. J Ayub Med Coll Abbottabad. 2011;23(1):102- 105.
- Mary JY, Baumelou E, Guiguet M. Epidemiology of aplastic anemia in France: a prospective multicentric study. The French ooperative Group for Epidemiological Study of Aplastic Anemia.Blood. 1990;75(8):1646- 53.
- Montané E, Ibáñez L, Vidal X, Ballarín E, Puig R, García N, Laporte J. Epidemiology Of Aplastic Anemia: A Prospective Multicenter Study. Haematologica 2008;93: 518-523
- Maluf EM, Pasquini R, Eluf JN, Kelly J, Kaufman DW. Aplastic anemia in Brazil: incidence and risk factors. Am J Hematol.2002;71(4):268-74.
- McCahon E, Tang K, Rogers PC, McBride ML, Schultz KR. The impact of Asian descent in the incidence of acquired severe aplastic anaemia in children. Br J Haematol. 2003;121(1):170-172.
- Yang C, Zhang X. Incidence survey of aplastic anemia in China. Chin Med Sci J. 1991;6(4):203-7.
- Kaufman DW, Kelly JP, Jurgelon JM, Anderson T, Issaragrisil S, Wiholm BE, Young NS, Leaverton P, Levy M, Shapiro S. Drugs in the aetiology of agranulocytosis and aplastic anaemia. Eur J Haematol Suppl. 1996;60:23-30.
- Yong AS, Goh AS, Rahman M, Menon J, Purushothaman V. Epidemiology of aplastic anaemia in the state of Sabah, Malaysia. Med J Malaysia. 1998;53(1):59-62.
- Ahamed M, Anand M, Kumar A, Siddiqui MK. Childhood aplastic anaemia in Lucknow, India: incidence, organochlorines in the blood and review of case reports following exposure to pesticides. Clin Biochem. 2006;39(7):762-766.
- Jacquemont C, Taniguchi T. The Fanconi anemia pathway and ubiquitin. BMC Biochemistry.2007;22(8): Suppl 1:S10.
- Alter BP, Giri N, Savage SA. Rosenberg PS. Cancer in dyskeratosis congenita.Blood.2009; 113:6549-6557.
- Shukla P, Ghosh K, Vundinti BR. Current and emerging therapeutic strategies for Fanconi anemia. Hugo J. 2012; 6(1): 1.
- Adam MP. Ardinger HH, Pagon RA. Wallace SE. Seattle (WA): University of Washington, Seattle, Gene Reviews; 1993-2018.
- Walne AJ, Vulliamy T, Beswick R, Kirwan M, Dokal I. TINF2 mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood. 2008;112(9):3594-600.
- Rujkijyanont P, Watanabe K, Ambekar C, Wang H, Schimmer A, Beyene J, Dror Y.SBDS-deficient cells undergo accelerated apoptosis through the Fas-pathway. Haematologica.2008;93(3):363-371.
- Clinton C, Gazda HT. Diamond-Blackfan Anemia. Gene Reviews [Internet]. Seattle (WA): University of Washington, Seattle;2009.1993-2017.
- Kagan WA, Ascensão JA, Pahwa RN, Hansen JA, Goldstein G, Valera EB, Incefy GS, Moore MA, Good RA. Aplastic anemia: presence in human bone marrow of cells that suppress myelopoiesis. Proc Natl Acad Sci U S A. 1976;73(8):2890-2894.
- Dutta A, De R, Dolai TK, Pal P, Ghosh S, Mitra PK, Halder A. Incidence of Fanconi anemia in phenotypically normal aplastic anemia patients in West Bengal. Hematology.2018;23(7):405-412.
- Young NS, Alter BP. Aplastic Anaemia: Acquired and Congenital. WB Saunders, Philadelphia. 1994
- Dutta A, De R, Dolai TK, Mitra PK, Halder A. Incidence of Fanconi Anemia in Morphologically Normal Aplastic Anemia Patients. Journal of the Vivekananda Institute of Medical Sciences. 2016;39(2):25-28.
- Dokal I. Dyskeratosis congenita in all its forms. British Journal of Haematology. 2000;110(4):768-779.
- Vulliamy T, Dokal I .Dyskeratosis congenita. Semin Hematol. 2006;43(3):157-166.
- Iwasaki T, Murakami M, Sugisaki C, Sobue S, Ohashi H, Asano H, Suzuki M, Nakamura S, Ito M, Murate T. Characterization of myelodysplastic syndrome and aplastic anemia by immunostaining of p53 and hemoglobin F and karyotype analysis: differential diagnosis between refractory anemia and aplastic anemia. Pathol Int. 2008;58(6):353-360.
- Appelbaum FR, Barrall J, Storb R, Ramberg R, Doney K, Sale GE, Thomas ED. Clonal cytogenetic abnormalities in patients with otherwise typical aplastic anemia. Exp Hematol. 1987;15(11):1134-1139.
- Alter BP.Diagnosis, genetics, and management of inherited bone marrow failure syndromes. Hematology Am Soc Hematol Educ Program. 2007:29-39.
- Vulliamy TJ, Walne A, Baskaradas A, Mason PJ, Marrone A, Dokal I. Mutations in the reverse transcriptase component of telomerase (TERT) in patients with bone marrow failure. Blood Cells Mol Dis. 2005;34(3):257-263.
- Dacie JV, Lewis SM, Practical Haematology, 9th edn. (ed. By S.M. Lewis, B.J. Bain & I. Bates), Churchill Livingstone,Elsevier, 2001,219–225.
- Socié G, Rosenfeld S, Frickhofen N, Gluckman E, Tichelli A. Late clonal diseases of treated aplastic anemia. Semin Hematol. 2000;37(1):91-101.
- Ishiyama K, Karasawa M, Miyawaki S, Ueda Y, Noda M, Wakita A, Sawanobori M, Nagai H, Nakao S. Aplastic anemia with 13q-: a benign subset of bone marrow failure responsive to immunosuppressive therapy. Br J Haematol. 2002;117(3):747-750.
- Maciejewski JP, Risitano A, Sloand EM, Nunez O, Young NS. Distinct clinical outcomes for cytogenetic abnormalities evolving from aplastic anemia.Blood. 2002;99(9):3129-3135.
- Sloand EM, Mainwaring L, Fuhrer M, Ramkissoon S, Risitano AM, Keyvanafar K, Lu J, Basu A, Barrett AJ, Young NS. Preferential suppression of trisomy 8 compared with normal hematopoietic cell growth by autologous lymphocytes in patients with trisomy 8 myelodysplastic syndrome. Blood. 2005;106(3):841-51.
- Sloand EM, Rezvani K, Mainwaring L, Barrett J, Mainwaring L, Kurlander R, Gostick E, Ramkissoon S, Tang Y, Douek D, Price D, Young NS, Myelodysplasia with trisomy 8 is associated with a cytotoxic CD8 T cell immune response to Wilms tumor-1 protein (WT1). Blood. 2004; 104: 138a. [Abstract 474]
- Kojima S, Ohara A, Tsuchida M, Kudoh T, Hanada R, Okimoto Y, Kaneko T, Takano T, Ikuta K, Tsukimoto I. Risk factors for evolution of acquired aplastic anemia into myelodysplastic syndrome and acute myeloid leukemia after immunosuppressive therapy in children. Blood. 2002;100(3):786-790.
- Sloand EM, Yong AS, Ramkissoon S, Solomou E, Bruno TC, Kim S, Fuhrer M, Kajigaya S, Barrett AJ, Young NS.Granulocyte colony-stimulating factor preferentially stimulates proliferation of monosomy 7 cells bearing the isoform IV receptor. Proc Natl Acad Sci U S A. 2006;103(39):14483-14488.
- Ohga S, Ohara A, Hibi S, Kojima S, Bessho F, Tsuchiya S, Ohshima Y, Yoshida N, Kashii Y, Nishimura S, Kawakami K, Nishikawa K and Tsukimoto Treatment responses of childhood aplastic anemia with chromosomal aberrations at diagnosis. Br J Haematol 2002; 118:313-319.
- Tichelli A, Gratwohl A, Würsch A, Nissen C, Speck B. Late haematological complications in severe aplastic anaemia. Br J Haematol. 1988;69(3):413-418.
- Mikhailova N, Sessarego M, Fugazza G, Caimo A, De Filippi S, Van Lint MT, Bregante S, Valeriani A, Mordini N, Lamparelli T, Gualandi F, Occhini D and Bacigalupo A. Cytogenetic abnormalities in patients with severe aplastic anemia. Haematologica. 1996;81(5):418-422.
- Geary CG, Harrison CJ, Philpott NJ, Hows JM, Gordon-Smith EC, Marsh JC. Abnormal cytogenetic clones in patients with aplastic anaemia: response to immunosuppressive therapy. Br J Haematol. 1999;104(2):271-274.
- Yamazaki E, Kanamori H, Taguchi J, Harano H, Mohri H, Okubo T. The evidence of clonal evolution with monosomy 7 in aplastic anemia following granulocyte colony-stimulating factor using the polymerase chain reaction. Blood Cells Mol Dis.1997;23(2):213-218.
- Gupta V, Kumar A, Saini I and Saxena AK. Cytogenetic profile of aplastic anaemia in Indian children. Indian J Med Res.2013; 137(3): 502- 506.
- Dutta A, De R, Dolai TK , Mitra PK, Halder A. Cytogenetic study is not essential in patients with aplastic anemia. Am J Blood Res. 2017;7(5):49-58.
- Brown KE, Tisdale J, Barrett AJ, Dunbar CE, Young NS. Hepatitis-associated aplastic anemia. N Engl J Med. 1997;336(15):1059– 1064.
- Oostra AB, Nieuwint AWM, Joenje H, de Winter JP. Diagnosis of Fanconi Anemia: Chromosomal Breakage Analysis. Hindawi Publishing Corporation. Volume 2012, Article ID 238731, 9 pgs.
- Jain D, Raina V, Fauzdar A, Mishra M, Tyagi N, Mahajan A, et al. Chromosomal breakage study in aplastic anemia patients in India. Asian J Med Sci.2010;2:227-232
- Grompe M, D’Andrea A. Fanconi anemia and DNA repair. Human Molecular Genetics. 2001;10(20):2253–2259.
- Palovcak A, Liu W, Yuan F, Zhang Y. Maintenance of genome stability by Fanconi anemia proteins. Cell Biosci. 2017;7:8.
- Nistico A, Young NS. Gamma-interferon gene expression in the bone marrow of patients with aplastic anemia. Ann Intern Med. 1994;120:463.
- Zoumbos NC, Gascon P, Djeu JY, Young NS. Interferon is a mediator of hematopoietic suppression in aplastic anemia in vitro and possibly in vivo. Proc Natl Acad Sci U S A. 1985;82(1):188-192.
- Gascon P, Zoumbos NC, Scala G, Djeu JY, Moore JG, Young NS. Lymphokine abnormalities in aplastic anemia: implications for the mechanism of action of antithymocyte globulin. Blood. 1985;65(2):407-413.
- Hinterberger W, Adolf G, Bettelheim P, Geissler K, Huber C, Irschick E , Kalhs P, Koller U, Lechner K, Meister B. Lymphokine overproduction in severe aplastic anemia is not related to blood transfusions. Blood.1989;74(8):2713-2717.
- Segel GB, Lichtman MA. Williams Hematology: Basic Principles and Practice.8th ed. McGraw-Hill Medical, New York: 2010, ch 34,463-481
- Feng X, Scheinberg P, Wu CO, Samsel L, Nunez O, Prince C, Ganetzky RD, McCoy JP (Jr), Maciejewski JP, Young NS. Cytokine signature profiles in acquired aplastic anemia and myelodysplastic syndromes. J Thromb Haemost.2012;10(8):1616-23.
- Feng X, Scheinberg P, Samsel L, Rios O, Chen J, McCoy JP Jr, Ghanima W, Bussel JB, Young NS.Decreased plasma cytokines are associated with low platelet counts in aplastic anemia and immune thrombocytopenic purpura. Clin Transplant. 1999;13(1 Pt 1):68-71.
- Sato S, Fuchinoue S, Abe M, Kitajima K, Tojimbara T, Nakajima I, Agishi T, Shiraga H, Ito K, Takasaki K, Hashimoto E, Hayashi N, Tanaka K. Successful cytokine treatment of aplastic anemia following living-related orthotopic liver transplantation for non-A, non-B, non-C hepatitis. Clin Transplant. 1999;13(1 Pt 1):68-71.
- Binder D, van den Broek MF, Kägi D, Bluethmann H, Fehr J, Hengartner H, Zinkernagel RM. Aplastic Anemia Rescued by Exhaustion of Cytokine-secreting CD8+ T Cells in Persistent Infection with Lymphocytic Choriomeningitis Virus. J Exp Med. 1998 ; 187(11): 1903–1920.
- Koike M, Ishiyama T, Tomoyasu S, Tsuruoka N. Spontaneous cytokine overproduction by peripheral blood mononuclear cells from patients with myelodysplastic syndromes and aplastic anemia. Leuk Res.1995;19(9):639-44.
- Zayed RA, Abdel-Hamid SM, El-Lithy H. The association of cytokine genes polymorphisms and susceptibility to aplastic anemia in Egyptian patients.Hematology. 2016; 21(2):106-112.
- Tripathy NK, Nityanand S; Vibhuti.Bone marrow and blood plasma levels of IL-8 in aplastic anemia and their relationship with disease severity. Am J Hematol.2005;79(3):240-2.
- De R, Dutta A, Dolai TK, Ghosh K, Halder A. Comparative study of bone marrow and blood plasma levels of IL-2 in aplastic anaemia and their relationship with disease severity. Hematology. 2018;23:1-5.
- Bacigalupo A. Aetiology of severe aplastic anaemia and outcome after allogeneic bone marrowtransplantationor immunosuppression. Eur J Haematol. 1996;57(60):16-19.
- Feng X, Chuhjo T, Sugimori C, Kotani T, Lu X, Takami A, Takamatsu H, Yamazaki H, Nakao S. Diazepam-binding inhibitor-related protein 1: a candidate autoantigen in acquired aplastic anemia patients harboring a minor populationofparoxysmalnocturnal hemoglobinuria-type cells. Blood. 2004;104(8):2425-2431.
- Ogawa S. Clonal hematopoiesis in acquired aplastic anemia.Blood. 2016;128(3):337-347.
- Kelsey P, Murphy MF, Brown M, Carrington P, Hall G, Jeffrey RR, Machin S, Taylor C, Thomas D. Guidelines for the use of platelet transfusions. Br J Haematol. 2003;122:10–23.
- Kaminski ER1, Hows JM, Goldman JM, Batchelor JR. Pretransfused patients with severe aplastic anaemia exhibit high numbers of cytotoxic T lymphocyte precursors probably directed at non-HLA antigens. Br J Haematol. 1990;76(3):401-405.
- Killick SB1, Win N, Marsh JC, Kaye T, Yandle A, Humphries C, Knowles SM, Gordon-Smith EC. Pilot study of HLA alloimmunization after transfusion with pre-storage leucodepleted blood products in aplastic anaemia.Br J Haematol. 1997 Jun;97(3):677-84.
- Pamphilon DH, Rider JR, Barbara JA, Williamson LM. Prevention of transfusion- transmitted cytomegalovirus infection. Transfus Med. 1999;9(2):115-123.
- Marsh JC1, Ganser A, Stadler M. Hematopoietic growth factors in the treatment of acquired bone marrow failure states. Semin Hematol. 2007;44(3):138-147.
- Casadevall N, Nataf J, Viron B, Kolta A, Kiladjian JJ, Martin-Dupont P, Michaud P, Papo T, Ugo V, Teyssandier I, Varet B, Mayeux P. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346(7):469-475.
- Schrezenmeier H, Hertenstein B, Wagner B, Raghavachar A, Heimpel H.A pathogenetic link between aplastic anemia and paroxysmal nocturnal hemoglobinuria is suggested by a high frequency of aplastic anemia patients with a deficiency of phosphatidylinositol glycan anchored proteins. Exp Hematol. 1995;23(1):81-87.
- Kurzrock R, Paquette R, Gratwohl A. Use of stem cell factor (Stemgen, SCF) and filgrastim (G-CSF) in aplastic anaemia patients who have failed ATG/ALG therapy. Blood.1997;90(Suppl. 1):173a.
- Vadhan-Raj S. Clinical experience with recombinant human thrombopoietin in chemotherapy-induced thrombocytopenia. Semin Hematol. 2000;37(2 Suppl 4):28-34.
- Bodey GP, Bolivar R, Fainstein V. Infectious complications in leukaemic patients. Seminars in Hematology.1982;19:193.
- Ljungman P. Supportive treatment of patients with severe aplastic anaemia.Aplastic Anaemia, Pathophysiology and Treatment (ed. by H. Schrezenmeier & A. Bacigalupo), Cambridge University Press, Cambridge, UK. 2000. 137-153.
- Porter JB. Practical management of iron overload.Br J Haematol. 2001;115(2):239-252.
- Hendry CL, Marsh JCW, Gordon-Smith EC, Sivakumaran M. Relapse of severe aplastic anaemia after influenza immunisation. . Br J Haematol.2002; 119: 268-288.
- Schrezenmeier H, Passweg JR, Marsh JC, Bacigalupo A, Bredeson CN, Bullorsky E, Camitta BM, Champlin RE, Gale RP, Fuhrer M, Klein JP, Locasciulli A, Oneto R, Schattenberg AV, Socie G, Eapen M.Worse outcome and more chronic GVHD with peripheral blood progenitor cells than bone marrow in HLA-matched sibling donor transplants for young patients with severe acquired aplastic anemia. Blood. 2007;110(4):1397-1400.
- Davies JK, Guinan EC.An update on the management of severe idiopathic aplastic anaemia in children. Br J Haematol. 2007;136(4):549-564.
- Niederwieser D, Pepe M, Storb R, Loughran TP Jr, Longton G. Improvement in rejection, engraftment rate and survival without increase in graft-versus-host disease by high marrow cell dose in patients transplanted for aplastic anaemia. Br J Haematol.1988;69(1):23-28.
- Gluckman E, Rocha V, Boyer-Chammard A, Locatelli F, Arcese W, Pasquini R, Ortega J, Souillet G, Ferreira E, Laporte JP, Fernandez M, Chastang C. Outcome of cord-blood transplantation from related and unrelated donors. Eurocord Transplant Group and the European Blood and Marrow Transplantation Group. N Engl J Med. 1997;337(6):373-381.
- Gupta V, Carreras J, Bajorunaite R, Gale RP, Sabloff M, Aljurf M, Schrezenmeier H, Socie´G, Passweg J, Ringde´n O, Pasquini R, Marsh J, Eapen M. Hematopoietic recovery and overall survival after HLA-matched sibling transplants for older patients with Severe Aplastic Anemia (SAA). Blood.2008. (ASH Annual Meeting Abstracts), 112, 2169.
- Lee SJ, Klein J, Haagenson M, Baxter-Lowe LA, Confer DL, Eapen M, Fernandez-Vina M, Flomenberg N, Horowitz M, Hurley CK, Noreen H, Oudshoorn M, Petersdorf E, Setterholm M, Spellman S, Weisdorf D, Williams TM, Anasetti C.High-resolution donor-recipient HLA matching contributes to the success of unrelated donor marrow transplantation. Blood. 2007;110(13):4576-4583.
- Bacigalupo A1, Brand R, Oneto R, Bruno B, Socié G, Passweg J, Locasciulli A, Van Lint MT, Tichelli A, McCann S, Marsh J, Ljungman P, Hows J, Marin P, Schrezenmeier H.Treatment of acquired severe aplastic anemia: bone marrow transplantation compared with immunosuppressive therapy-- The European Group for Blood and Marrow Transplantation experience. Semin Hematol. 2000;37(1):69-80.
- Ades L, Mary JY, Robin M, Ferry C, Porcher R, Esperou H, Ribaud P, Devergie A, Traineau R, Gluckman E, Socie G. Long-term outcome after bone marrow transplantation for severe aplastic anaemia. Blood. 2004;103(7):2490– 2497.
- Rosenberg PS, Alter BP, Socié G, Gluckman E. Secular trends in outcomes for Fanconi anemia patients who receive transplants: implications for future studies. Biol Blood Marrow Transplant. 2005;11(9):672-679.
- Schrezenmeier, H., Bacigalupo, A., Aglietta, M., Camitta, B., Frickhofen, N., Fuhrer, M., Gluckamn, E., Gratwohl, A., Heimpel, H., Hows, J., Kojima, S., Locasciulli, A., Marmont, A., Marin, P., Marsh, J., McCann, S., Passweg, J., Pasquini, R., Podesta, M., Socie, G., Storb, R., Tichelli, A., Torok-Storb, B., Wodnat-Filipovicz, A. & Young, N. Guidelines for treating aplastic anemia: consensus document of a group of international experts. Aplastic Anemia-Pathophysiology and Treatment. Cambride University Press, 2000, 308-315.
- Maury S, Viollier R, Oneto R, Anderlini P, Aljurf M, Garban F, Cordonnier C, Marsh J, Bacigalupo A, Passweg J. Overcoming the negative impact of age using fludarabine based conditioning regimens for HLA-identical sibling HSCT in patients with severe aplastic anaemia. Bone Marrow Transplantation.39(S1).Abstract 327.
- Lawler M, McCann SR, Marsh JC, Ljungman P, Hows J, Vandenberghe E, O'Riordan J, Locasciulli A, Socié G, Kelly A, Schrezenmeier H, Marin P, Tichelli A, Passweg JR, Dickenson A, Ryan J, Bacigalupo A. Serial chimerism analyses indicate that mixed haemopoietic chimerism influences the probability of graft rejection and disease recurrence following allogeneic stem cell transplantation (SCT) for severe aplastic anaemia (SAA): indication for routine assessment of chimerism post SCT for SAA. Br J Haematol. 2009 Mar;144(6):933-45.
- McCann SR, Passweg J, Bacigalupo A, Locasciulli A, Locatelli F, Ryan J, Schrezenmeier H, Lawler M. The influence of cyclosporin alone, or cyclosporin and methotrexate, on the incidence of mixed haematopoietic chimaerism following allogeneic sibling bone marrow transplantation for severe aplastic anaemia. Bone Marrow Transplant. 2007;39(2):109-114.
- Dubey S, Nityanand S. Involvement of Fas and TNF pathways in the induction of apoptosis of T cells by antithymocyte globulin. Ann Hematol. 2003;82(8):496-499.
- Michallet MC, Saltel F, Preville X, Flacher M, Revillard JP, Genestier L. Cathepsin-B- dependent apoptosis triggered by antithymocyte globulins: a novel mechanism of T-cell depletion. Blood. 2003;102(10):3719-26.
- Mangan KF, D'Alessandro L, Mullaney MT. Action of antithymocyte globulin on normal human erythroid progenitor cell proliferation in vitro: erythropoietic growth-enhancing factors are released from marrow accessory cells. J Lab Clin Med. 1986;107(4):353-364.267.
- Bielory L, Wright R, Nienhuis AW, Young NS, Kaliner MA. Antithymocyte globulin hypersensitivity in bone marrow failure patients.JAMA. 1988;260(21):3164-3167.
- Marsh J, Schrezenmeier H, Marin P, Ilhan O, Ljungman P, McCann S, Socie G, Tichelli A, Passweg J, Hows J, Raghavachar A, Locasciulli A, Bacigalupo A. Prospective randomized multicenter study comparing cyclosporin alone versus the combination of antithymocyte globulin and cyclosporin for treatment of patients with nonsevere aplastic anemia: a report from the European Blood and Marrow Transplant (EBMT) Severe Aplastic Anaemia Working Party. Blood. 1999;93(7):2191-2195.
- Tichelli A, Socié G, Henry-Amar M, Marsh J, Passweg J, Schrezenmeier H, McCann S, Hows J, Ljungman P, Marin P, Raghavachar A, Locasciulli A, Gratwohl A, Bacigalupo A. Effectiveness of immunosuppressive therapy in older patients with aplastic anemia. European Group for Blood and Marrow Transplantation Severe Aplastic Anaemia Working Party. Ann Intern Med. 1999; 130(3):193-201.
- Kao SY, Xu W, Brandwein JM, Lipton JH, Messner HA, Minden MD, Schimmer AD, Schuh AC, Yee K, Gupta V. Outcomes of older patients (> or = 60 years) with acquired aplastic anaemia treated with immunosuppressive therapy. Br J Haematol. 2008;143(5):738-743.
- Hill A, Hillmen P, Richards SJ, Elebute D, Marsh JC, Chan J, Mojcik CF, Rother RP. Sustained response and long-term safety of eculizumabinparoxysmalnocturnal hemoglobinuria.Blood. 2005;106(7):2559-2565.
- Saso R, Marsh J, Cevreska L, Szer J, Gale RP, Rowlings PA, Passweg JR, Nugent ML, Luzzatto L, Horowitz MM, Gordon-Smith EC.Bone marrow transplants for paroxysmal nocturnal haemoglobinuria. Br J Haematol. 1999;104(2):392-396.
- Hillmen P, Muus P, Dührsen U, Risitano AM, Schubert J, Luzzatto L, Schrezenmeier H, Szer J, Brodsky RA, Hill A, Socié G, Bessler M, Rollins SA, Bell L, Rother RP, Young NS. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria.Blood. 2007;110(12):4123-4128.
- Hall C, Richards S, Hillmen P. Primary prophylaxis with warfarin prevents thrombosis in paroxysmal nocturnal hemoglobinuria (PNH). Blood. 2003;102(10):3587-3591.
- Oosterkamp HM, Brand A, Kluin-Nelemans JC, Vandenbroucke JP.Pregnancy and severe aplastic anaemia: causal relation or coincidence?.Br J Haematol. 1998;103(2):315-316.
- Sanders JE, Hawley J, Levy W, Gooley T, Buckner CD, Deeg HJ, Doney K, Storb R, Sullivan K, Witherspoon R, Appelbaum FR. Pregnancies following high-dose cyclophosphamide with or without high-dose busulfan or total-body irradiation and bone marrow transplantation. Blood.1996;87(7):3045-3052.
- Provan D, Newland AN, Norfolk D, Bolton-Maggs P, Lilleyman J, Greer I, May A, Murphy M, Ouwehand W, Watson S. Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol. 2003;120(4):574-96.