


|
|
DOI Prefix 10.20431 |
Information
Journal Policies
Detection of Homozygous Haemoglobin Constant Spring by Capillary Electrophoresis Method
Raja-Zahratul Azma1*, Khamisah M-Gaus1, Suria A-Aziz1, Hafiza Alauddin1, Azlin Ithnin1, Noor-Farisah Razak1, Nor-Hidayati Sardi1, Malisa Mohd-Yusoff1, Zarina A-Latiff2, Hamidah Alias2, Endom Ismail3, Danny Koh-Xuan-Rong3, Ainoon Othman4
2 Paediatric UKM Medical Centre, Kuala Lumpur.
3 School of Bioscience and BiotechnologyFaculty of Science and Technology University Kebangsaan Malaysia, Bangi, Malaysia.
4 Faculty of Medicine and Health Sciences, University Sains Islam Malaysia (USIM) Kuala Lumpur, Malaysia.
Copyright : © 2016 Khojasteh HN. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Introduction: Hemoglobin Constant Spring (HbCS) is often missed due to almost normal red cells indices and is present at a low level in peripheral blood. However, analysis of blood samples using capillary electrophoresis (CE) is able to differentiate between heterozygous and homozygous state of HbCS.
Methods: An eight-year-old Malay girl presented with severe anaemia and hepatosplenomegaly following a viral infection. The diagnosis of homozygous HbCS was made from Hb analysis by CE which showed a small peak (5.3%) at Hb CS zone. His brother had similar CE findings while both of her father and mother showed much smaller peaks (0.7% and 0.4% respectively) at similar zone which were consistent with heterozygous HbCS. All findings were confirmed by few molecular analysis techniques.
Conclusion: This case highlights the importance of detection of homozygous HbCS by CE. CE is useful as a fast screening method for detection of HbCS in hematology laboratory where molecular analysis is not available and patient with homozygous HbCS has higher risk to develope acute hemolysis when having infection.
Keywords: Capillary Electrophoresis, Hb Constant Spring, Homozygous State
1.Introduction
HbCS is the most prevalent non-deletional alpha (α) thalassaemia in the South East Asia population. In Malaysia, the frequency is higher in Malays (2.24%) compared to Chinese and Indian (0.66% and 0.16% respectively)[1]. HbCS is formed as a result of a mutation in the stop codon of the α2-globin gene (TAA>CAA) that leads to reduction in α chain synthesis (1% of normal) which now has 31 additional amino acids[2]. The unstable nature of HbCS may result in a missed detection by methods usually employed in Malaysian laboratories. Hb analysis using fresh blood samples by cellulose acetate electrophoresis at alkaline pH can easily identify HbCS band particularly if a heavy application is used. The band moves between carbonic anhydrase enzyme and Hb A2 while in High Performance Liquid Chromatography (HPLC) it appears in the C window. However, in our previous experience[3] sometimes there was no peak seen in this region for heterozygotes HbCS but, presence of HbCS was noted in Capillary Electrophoresis (CE) whereby the peak of this Hb was detected at HbC/CS zone. Our findings were consistent with a study done by Waneesorn et al.[4] which showed that the CE method was superior to HPLC method in detecting HbCS especially in heterozygote state. Molecular analysis is a definitive method for diagnosis of HbCS. The usual molecular technique used in Malaysian laboratories for the identification of α-globin gene mutations is multiplex amplified refractory mutation system (ARMS) polymerase chain reaction (PCR). However this molecular method is not available in most of laboratories in Malaysia and need duplicated runs before we can differentiate between homozygous and heterozygous state of HbCS. DNA analysis using real time PCR using Taqman@ SNP genotyping assays is able to differentiate between homozygous and heterozygous state of HbCS, but this molecular test is expensive and laborious
2. Case Report
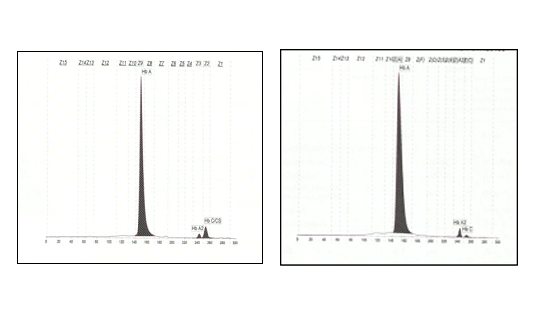
We present our experience in diagnosing a case of Homozygous HbCS using the CE. The patient was an eight-year-old Malay girl who presented with symptomatic anaemia and hepatosplenomegaly following viral infection. The patient is the eldest of two siblings. Her haematology profiles were: Hb 6.8 g/dL, RCC 2.96 X 1012/L, MCV 76.6 fl, MCH 22.6 pg, MCHC 29.8 g/dL, RDW 15%, WCC 6.9 X 109/L and platelet count 334 x 109/L. Peripheral blood film showed hypochromic microcytic red cells with anisocytosis and leucoerythroblastic picture. There were features of haemolysis such basophilic stippling and polychromasia. Both reticulocytes count and serum lactate dehydrogenase were raised. Her Hb analysis by CE showed normal HbA2 and HbF levels with an additional small peak (5.3%) at Hb C/CS zone (Figure 1)
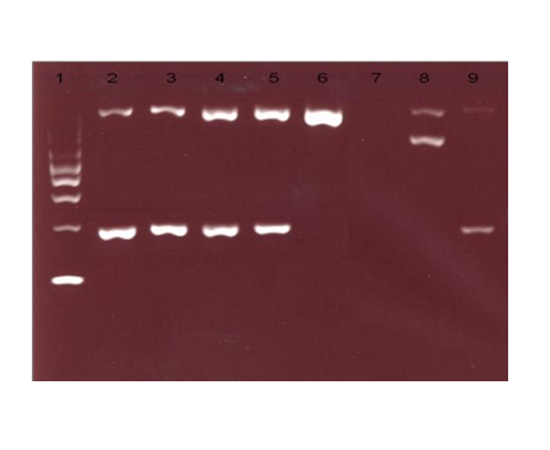
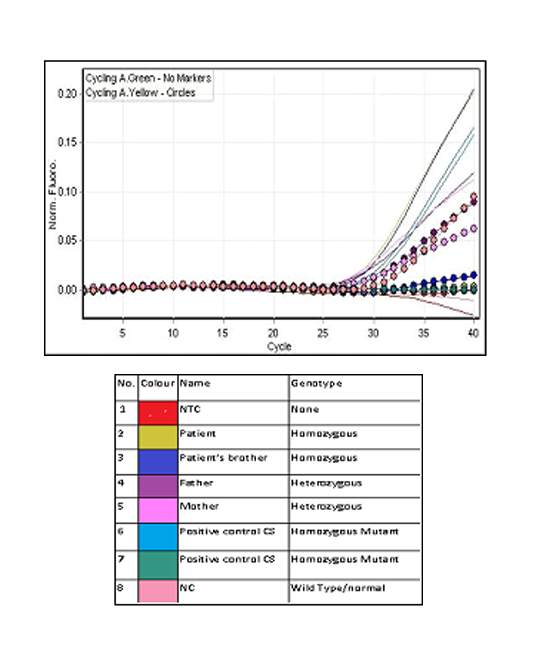
While HPLC showed presence of a small peak(3.9%)at C-window. DNA analysis by multiplex ARMS PCR confirmed the presence of HbCS (Figure 2) and real time PCR using Taqman@ SNP genotyping assays confirmed that the patient is homozygous for Hb CS (Figure 3).
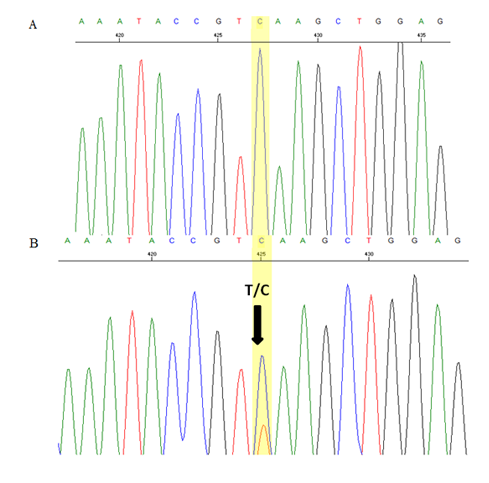
Blood samples from her family members were also analysed. Her parents had normal Hb levels with mild hypochromic microcytic red cells indices and the CE also showed small peaks (0.4% and 0.7%) at HbCS zone. There was a small peak (0.6%) at C-window seen in HPLC of her father’s blood sample but no similar peak seen in her mother’s blood sample. Her three-year-old brother had almost similar laboratory findings to her except for moderate anaemia with Hb of 9.7 g/dL. DNA analysis using real time PCR using Taqman@ SNP genotyping assays confirmed that both of her parents are heterozygous for HbCS, while his brother is also homozygous for HbCS (Figure 3). Sequencing analysis on the HBA2 gene of the patient and her brother confirmed that they are HbCS homozygotes while their parents are HBCS heterozygotes (Figure 4).
The patient was transfused with leucocytes poor red blood cells in view of decreasing trend of haemoglobin level to 5.0 x 109 g/dL. She remained transfusion free after discharge until the present time. Her latest Hb was 10.7 x 109 g/dL.
3. Discussion
Homozygous HbCS is usually asymptomatic as shown by this patient who remained undiagnosed until at the age of 8 years old when she presented with symptomatic anaemia following viral infections. Acute haemolysis associated with mild upper respiratory tract infection has been reported in a Thai boy with homozygous HbCS who presented with a rapid decline in haemoglobin (Hb) levels, haemoglobinuria and evidence of intravascular haemolysis[5]. Peripheral red blood cells (RBCs) destruction appears to account for anaemia in severe α-thalassaemia as opposed to ineffective eryhtropoeisis in β-thalassaemia. Accumulation of β globin chains in severe α thalassaemia results in increased hydration of RBCs and in addition to presence of αCS would have deleterious effects on RBCs which lead to more severe haemolysis[6].
The patient had a severe haemoglobin level at presentation as compared to her brother, which was secondary to the acute haemolysis and remained stable at 10 g/dL after four months without blood transfusion. The patient, her brother and her mother had low MCH with borderline low MCV. However both her father had normal red cells parameters. Heterozygotes of HbCS usually have a normal Hb level with normal MCV and slightly low MCH[3] however, similar findings were not seen in her mother. In HbCS, the reduction of the MCH is not so marked with normal mean MCV due to damage of red cell membranes by oxidized α and αCS globin chains which leads to cellular overhydration[7]. Similar findings were seen in both of the patient’s parents. Thus carrier states of HbCS could be easily missed if the screening is mainly based on red cell indices.
Her diagnosis of HbCS was confirmed by multiplex ARMS PCR. However, in view of her presentation with acute haemolysis with severe anaemia and hepatosplenomegaly, thalassaemia intermedia or unstable haemoglobin disease needed to be considered. Her blood investigations showed HbCS of 2.9% by HPLC and 5.3% by CE and with the family studies of both parents who are positive for HbCS, these supported the diagnosis of homozygous HbCS. Homozygous HbCS is one of the genetic abnormalities leading to HbH disease[7]. Clinical features of HbH disease varies from a mild disease, in which the diagnosis is made incidentally, to a much more severe anaemia that requires intermittent or rarely regular blood transfusions. The anaemia is aggravated by infections, pregnancy or exposure to oxidant drugs, and can be worsened by superimposed folate deficiency or aplastic crisis. The incidence of gallstones and cholecystitis is increased among this group[7]. A case of acute haemolysis associated with mild upper respiratory tract infection was observed a Thai boy with Homozygous HbCS[5].
The definitive diagnosis of HbCS requires DNA analysis. The abnormal α globin is unstable, thus the fraction of this slow moving haemoglobin can be missed when non-molecular techniques are used. The variant haemoglobin may be detected by haemoglobin electrophoresis or HPLC, but it comprises a very low proportion of total haemoglobin. HbCS can be identified on cellulose acetate electrophoresis at alkaline pH, particularly if a heavy application is used where it moves between carbonic anhydrase and HbA2[7]. At acid electrophoresis, it moves together with Hb S. On HPLC it appears in the C-window. In this patient’s family study, the gel electrophoresis showed no abnormal band except for the patient’s brother that showed presence of slow band between carbonic anhydrase and HbA2 at alkaline pH and abnormal band appeared in S region at acid HbCS was detected by HPLC in all three family members except for the mother’s sample. In contrast, CE detected the presence of HbCS in all the family members, with a lower level (0.4% and 0.7%) of HbCS in heterozygotes compared to homozygotes (4.7% and 5.3%). Study by Waneesorn et al.[4] concluded that CE was superior to HPLC for detecting Hb CS. In their study, HbCS peaks were found in 26.32% heterozygous, 42.86% homozygous and 90% HbH-CS by HPLC and were detected in all samples by CE. They also concluded that the level of HbCS quantified by CE is useful in screening heterozygous and homozygous CS as well as HbH-CS. Similar study by Liao et al.8 also concluded that CE is the preferred method for HbCS screening.
Multiplex-ARMS PCR used for the detection of HbCS in our centre requires an additional run to look for presence of normal α2 allele (wild type) before it can distinguish between homozygous and heterozygous HbCS. However, correlation with clinical and haematological findings together with HbCS level by CE will raised a suspicion towards homozygous HbCS or compound heterozygous with some other haemoglobin variant. The detection of common α gene deletions were also carried out in this family as compound heterozygous with two gene deletion could present with a similar picture. Homozygous HbCS can be demonstrated by DNA analysis using real-time PCR. In this technique, amplification of the mutant allele for HbCS and normal α2 allele (wild type) will be carried out using allele-specific fluorescence PCR[3]. In homozygous HbCS, only mutant allele will be amplified without amplification of the wild type allele. Tangvarasittichai et al.[9] demonstrated that DNA based method, restriction enzyme digestion (RED) and ARMS, were efficient diagnostic tools in detecting HbCS in homozygous and heterozygous. In RED, amplified DNA of the α2 globin gene encompassing the terminal codon was digested with MseI. MseI cuts DNA at restriction site TTAA to produce two fragments of 165bp and 111bp in normal individuals, whereas undigested 276bp amplicon will remain in individual with HbCS. However, RED could not distinguish between HbCS and other α2 globin gene terminal mutations such as HbCSPakse. Thus, ARMS is suggested as a confirmative diagnosis and mutation cases unresolved by ARMS should be clarified by appropriate methods such as DNA sequencing.
In terms of clinical management, HbCS results in a mild form of alpha thalassaemia and does not require specific treatment. Occasional red blood cells transfusions may be needed if the haemoglobin level suddenly drops due to acute haemolytic crisis, secondary to viral infections as seen in this patient. Folic acid supplementation may be beneficial in patients with elevated reticulocyte counts, indicating increased utilization as a result of the hemolyitic process and a high bone marrow turnover rate. Regular transfusion is indicated for thalassaemia patients when there is growth failure, bone marrow deformities or extramedullary masses (CPG-Management of Transfusion Dependent Thalassaemia).
In summary, this case has highlighted the ability of CE in detecting homozygous HbCS. This method can be used as a screening tool for detection of this Hb variant which is particularly relevant in Malaysian populations where this abnormal haemoglobin is highly prevalent. It also highlights the importance of homozygous HbCS as a potential predisposing cause of acute haemolysis in children that might be aggravated by acute bacterial or viral infections.
4. Acknowledgements
We would like to thank Puan Nur-Ahlina Abdullah and Muhd-Firdaus Rosli in helping us with the manuscript.
References
- Wee YC, Tan KL, Chow TW, Yap SF, Tan JA. Heterogeneity in α thalassaemia interactions in Malays, Chinese and Indians in Malaysia. J Obstet Gyanaecol Res. 2005; 31: 540-6.
- Suthat F, Pranee W. Haemoglobinopathies in Southeast Asia. Indian J Med Res. 2011; 134: 498– 506
- Azma RZ, Othman A, Azman N, Alauddin H, Ithnin A, Yusof N et al. Co-inheritance of compound heterozygous Hb Constant Spring and a single – α3.7 gene deletion with heterozygous δβ thalassaemia: A diagnostic challenge. Malaysian J Pathol. 2012; 34(1): 57-62.
- Waneesorn J, Panyasai S, Kongthai K, Singboottra P, Pornprasert S. Comparison between capillary electrophoresis and high performance liquid chromatography for detection and quantification of Hb Constant Spring. Hemoglobin. 2011; 35(4): 338-45.
- Viprakasit V, Veerakul G, Sanpakit K, Pongtanakul B, Chinchang W, Tanphaichitr VS. Acute haemolysis in a Thai patient with homozygous Hb Constant Spring: a case report. Ann Trop Paediatr. 2004; 24(4): 323-8.
- Bunyaratvej A, Fucharoen S, Greenbaum A, Mohandas N. Hydration of red cells in α and β thalassaemias differs. Am J Clin Pathol. 1994; 102(2): 217-22.
- Bain BJ. 2006. Haemoglobinopathy Diagnosis, 2nd ed. Wiley Blackwell, Oxford, 2006.
- Liao C., Zhou JY, Xie XM, Li J, Li R, Li DZ. Detection of Hb Constant Spring by a capillary electrophoresis method. Hemoglobin. 2010; 34(2): 175-8.
- Tangvarasittichai O, Jeenapongsa R, Sitthiworanan C, Sanguansermsri T. Laboratory investigations of Hb Constant Spring. Clin Lab Haematol. 2005; 27(1): 47-9.