


|
|
DOI Prefix 10.20431 |
Information
Journal Policies
Successful PCR using Old Conservation Museum Fluid is Possible - Preliminary Studies on the ABO Group Model
Malgorzata Malodobra-Mazur1*, Mike Preisner2, Malgorzata Bonar1, Matylda Czosnykowska-Lukacka1, Anna Jonkisz1, Mariola Turkiewicz1, Natalia Winiarska1, Tadeusz Dobosz1
2.University of Wolverhampton, School of Applied Sciences, Wolverhampton, United Kingdom
Copyright :© 2018 Authors. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Museum specimens are valuable resources for the study of diverse histological processes. Ancient DNA from museum specimens is enormously important in medical, phylogenetic and population genetic analysis. Unfortunately, most common DNA extraction methods rely on tissue degradation prior to DNA isolation, thereby entailing the destruction of a valuable specimen. However, application of non-destructive approaches could help to obtain DNA suitable for downstream applications such as PCR and to preserve tissue integrity for future use. In this paper we present a non-destructive method for DNA extraction from museum specimens stored in liquid media. Human tissue, filter paper used to filter conservation fluid and dilated, dialyzed conservation fluids were used for DNA extraction. The results showed that the same replicable PCR profile was derived from all three sources of the single sample, indicating that this is a viable method for the non-destructive retrieval of DNA from museum specimens preserved in liquid media.
Keywords: Museum specimens, Non-destructive DNA extraction, Ancient DNA, Multiplex PCR, ABO,Forensic Science
1.Introduction
Museum specimens are indubitably a valuable resource for studying diverse histological processes. Ancient DNA (aDNA) offers the potential for investigation of evolutionary changes over time and is enormously important in phylogenetic and population genetic studies (Bi 2013; Wandeler 2007). It also affords a rare insight into human history and the ambient world. However, museum specimens are difficult to analyze because they typically contain short fragments of denatured and often degraded DNA. For instance, DNA extraction is the first step in numerous molecular biology experiments, but most common DNA extraction methods rely on tissue degradation prior to DNA purification, which is less than ideal when handling most specimens from museums, where curators need to preserve material indefinitely. Although some museums approve the destructive analysis of small parts of their specimens, this does not apply to extremely valuable and unique samples.
Numerous non-destructive DNA extraction methods have been developed. Initially non-destructive methods were developed to extract mitochondrial DNA (mtDNA) only, as it is present in higher copy number in cells than genomic DNA (Tin 2014; Rohland 2004). Subsequent studies (Bolnik 2012) improved the method created by Rohland and Hofreiter (2007), which enabled amplification of nuclear DNA; however, only relatively short amplicons with approximately 250 bp (base pairs) were retrieved.
Most of the non-destructive protocols developed to date are designed mainly for aDNA extraction from skeletal remains, bones and teeth (Rohland 2004; Bolnik 2012; Rohland 2007; Hervella 2012). Like solid tissue, DNA suitable for genetic studies can be retrieved from bone, while simultaneously preserving the overall morphology of the tissue.
In cases of soft tissues such as human organs and tumor tissue, most of the available methods for DNA extraction require tissue degradation, generally by means of proteinase K digestion in a various detergent-based buffers, followed by DNA purification (Yang 1997; Bonin 2010; Paireder 2013).
In this article, we aimed at isolating PCR-amplifiable DNA from soft museum specimens stored in liquid media by applying a non-destructive method. We extracted DNA from museum specimen-related materials, including tissue itself, conservation fluid, and the filter paper used to filter conservation fluid. We proved that the DNA obtained from these materials works in PCR despite considerable DNA degradation.
2. Material And Methods
The study protocols were approved by ethical review boards at the Wroclaw Medical University (No: KB-475/2017).
DNA extraction, PCR set-up and post-PCR applications were carried out in a separate laboratory rooms. The extraction laboratory room was situated in a different building than the PCR and post-PCR lab. Disposable laboratory equipment was used throughout the analyses. Workspaces and equipment were decontaminated regularly. The laboratory rooms and laminar-flow hoods were UV irradiated for 1 hour before each use. Extraction controls were performed for both filter paper and conservation fluid. Reagents for amplifications were frozen as a aliquots, and new aliquots were taken every time PCR was set up. The negative controls (no template controls NTC) were performed for each PCR setup.
In affiliation with our local transfusiology unit, anonymized bloodstain samples of known ABO/RhD blood groups from Polish Caucasian volunteers were prepared. Blood stains collected from individuals were used as controls to validate the multiplex PCR system (ABO/amelogenin). Prior to the collection of blood samples all volunteers gave written consent to ABO and Rh determination, safety analysis, and medical usage.
Determination of ABO was carried out using the classical serology method prior to analysis of blood samples using the molecular approaches (ABO/amelogenin PCR assay). The bloodstains were prepared by pouring fresh whole blood obtained from cubical veins into filter paper and drying at room temperature, followed by storage at room temperature inside paper envelopes.
Pieces approximately 2mm2 were cut from the bloodstains and placed into 1.5 ml Eppendorf tubes, each containing 1 ml of distilled water to incubate at room temperature for 15 to 30 min. Solutions were centrifuged at 13,500 rpm for 3 min. The supernatant was removed and discarded; then 180 µl of a 5% Chelex suspension was added to each tube with the bloodstain and incubated at 56°C for 20 min. Next, tubes were incubated for 8 min at 98°C and centrifuged at 13,500 rpm for 5 min. Extracted DNA, along with the Chelex suspension was stored at 4°C for up to two weeks. Only the upper part of sample was used for PCR.
The collection of museum specimens came from the Museum Gerichtsärtzliches Institut D KCL Universität Breslau and was created before Second World War; it currently belongs to the Museum of the Cathedral of Forensic Medicine at Wroclaw Medical University, and represents various tissues of the human body. Documentation such as catalogue numbers or numerical assignments was completely lacking for the museum specimen collection. The dates and contents of the jars were recorded based on the description labels on each jar. The samples covered a period of over 150 years. The tissue samples were kept in their original jars, at room temperature in low light. For DNA extraction, aside from human tissues, the dialyzed conservation fluid and filter paper (Whatman GF/C) used for conservation fluid filtration were utilized. The museum conservation fluid was composed of water, ethanol (30%) and glycerol (10%). In the present study eight tissue samples, eight filter papers used for filtration, and 20 samples of conservation fluid were tested. Due to low income of positive results for conservation fluid regarding long amplicons, we increased the number of analyzed samples to strengthen the power of statistic analysis.
The conservation fluid was filtered through GF Whatman filter using vacuum pump. The filtered conservation fluid (50 µl) was pipetted into Visking Dialysis Tube (membrane) with a diameter of 8 mm and dialyzed at 4°C in a magnetic stirrer for 24 h in a PCR buffer (100 mM Tris, 500 mM KCl, 0.1% (v/v) Tween 20 and 2 mM magnesium chloride, adjusted to pH 8.35 by HCl) and for 12 h in deionized water; following dialysis, 4 µl of the preparation was added to the PCR mix prior to amplification. The concentration of DNA was measured using Qubit Fluorometer (Invitrogen).
A 25 mg portion of the tissue sample or 1 cm2 of the filter paper used for the filtration of the conservation fluid was cut into small pieces and placed into a micro-centrifuge tube, to which 180 µl of Buffer T1 and 25 µl of Proteinase K solution were added. It was then vortexed and incubated for 90 min at 56 °C in a shaking incubator set at 600 rpm until complete lysis of the tissue has been accomplished. The next steps of preparation were done in accordance with the manufacturer’s instructions (Macherey-Nagel). The obtained DNA samples were frozen at - 20 °C.
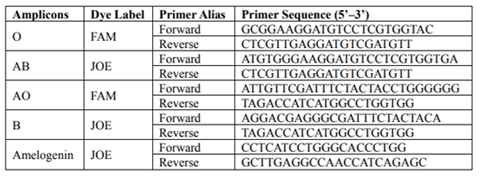
The sequences of the ABO primers (Jiang 2012) along with the fluorescent dyes are presented in Table 1. Two fluorescent dyes were applied (FAM and JOE) and labeled at the 5’ end of the forward primers. The obtained products ranged from 50 to 100bp and did not overlap.
A longer amplicon was added to the ABO assay to validate the rate of DNA degradation. The selected polymorphic locus was amelogenin, flanking a six-nucleotide deletion polymorphism, giving a 209 bp PCR product for the female and 209/215 bp for the male. The sequences of amelogenin primers, along with fluorescent dye, are presented in Table 1.
The PCR consisted of a 2 x Qiagen® Multiplex PCR Kit (Qiagen), 0.16 µM of each primer, and 4 µl of DNA, and was filled up to 10 µl with DNase, RNase-free water (Qiagen). Amplification was performed using a BioRad™ S1000 thermal cycler under the following conditions: (95 °C, 15 min); (94 °C, 30 s; 63 °C, 60 s; 72 °C, 45 s) x 30; (72 °C, 10 min); 4 °C indefinitely.
A ROX500 Internal Size Standard was used in accordance with the manufacturer’s instructions (Applied Biosystems). The PCR product (1 µl) was added to 10 µl of HiDi Formamide containing the ROX500 Internal Size Standard. The samples were denatured using a GeneAmp® 9700 (Applied BioSystems) at 95°C for 5 min and snap frozen at 0°C for 3 min. Samples were separated on an ABI Prism 3130 Genetic Analyzer using POP-4 Polymer.
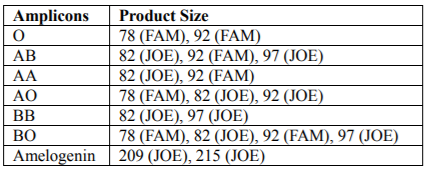
The product sizes obtained by multiplex-PCR did not overlap and were easily distinguished. Depending on the ABO blood group up to four PCR products (78 / 82 /92/97bp) were obtained, according to the pattern presented in Table 2. The B group gave two or four PCR products: two were seen in BB homozygotes (82/97 bp) and four in BO heterozygotes (78/82/92/97 bp). Similarly, the A group gave two or three PCR products: 82 and 92 bp in AA homozygotes and three (78/82/92 bp) in AO heterozygotes. The presence of two products (78/92 bp) corresponded to the O group. The AB genotype was characterized by the acquisition of three PCR products 82, 92 and 97 bp in length. The products of the amelogenin gene were as follow: male was represented by two signals of 209 and 215 bp, while female was represented by one signal of 209 bp. The size of the ABO and amelogenin alleles are shown in Table 2.
3. Results
We expected that DNA extracted from museum specimens would be partially degraded; therefore, we decided to test extracted DNA using a PCR-based ABO genotyping assay yielding relatively small amplicons, below 100 bp. Additionally, a longer amplicon (amelogenin) was amplified in multiplex PCR to estimate the extent of DNA degradation (that is, to determined whether fragments longer than 200 bp might be amplified).
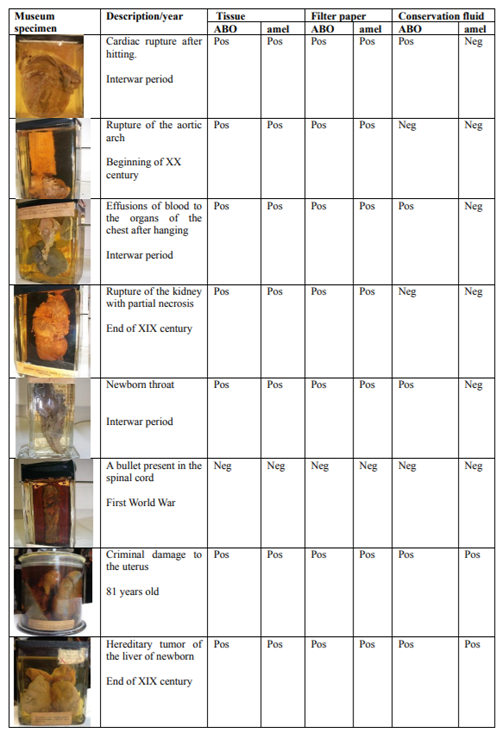
In order to validate the PCR conditions and to investigate whether the ABO genotypes corresponded to the ABO blood groups determined by the serological method, modern bloodstain samples were used. DNA extracted from bloodstains was amplified and the ABO results compared with the blood group diagnosed using the serological method. In all tested samples, the ABO genotype corresponded to the ABO blood group determined via the classical serological method.
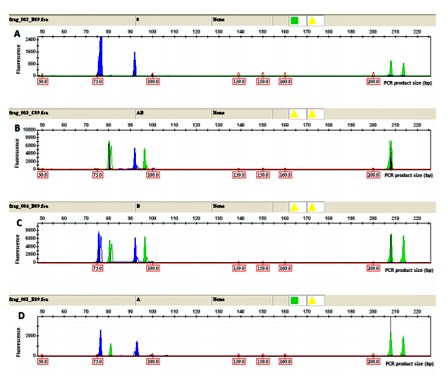
Examples of electropherograms showing ABO alleles O, AB, BO and AO and amelogenin genotypes from bloodstains are presented in Figure 1.
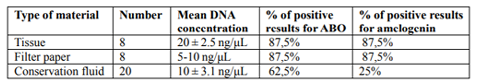
DNA from tissues and filter papers was isolated using the spin column method. The filtered conservation fluid was dialyzed at 4°C and directly subjected to further analysis. Twenty-four DNA samples (extracted from the set of 8 museum specimens) were used for the study. Additionally, more, not related conservation fluid samples were analyzed. The mean DNA concentrations obtained from human tissue, filter papers and conservation fluids were 20 ± 2.5 ng/μl, 5-10 ng/μl and 10 ± 3.1 ng/μl, respectively.
Following the optimization of PCR conditions, the ABO assay was used to test the DNA extracted from the museum specimens.
The whole profile of both ABO and amelogenin markers were obtained when DNA extracted from tissue and filter paper was used. Positive results were obtained for 90%. Similarly, the DNA extracted from dialyzed filtered conservation fluid was found to provide interpretable results. ABO PCR products were detected in 60% of the DNA extracts from conservation fluid, with approximately 25% producing full profiles for ABO and amelogenin (Figure 2). This result suggests that the DNA in conservation fluid, was more degraded than the DNA in tissue and filter paper, as longer PCR products failed to be amplified in the majority of cases. However, short amplicons up to 100 bp were successfully amplified more than half of the time (Table 3, Table 4).
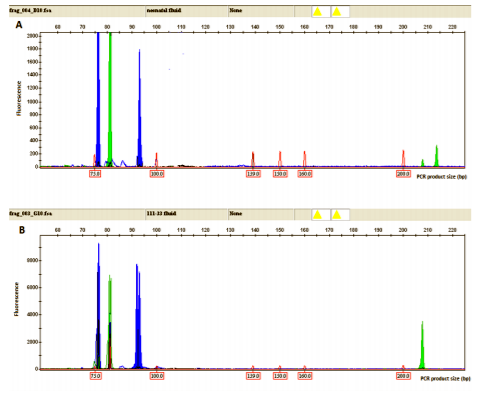
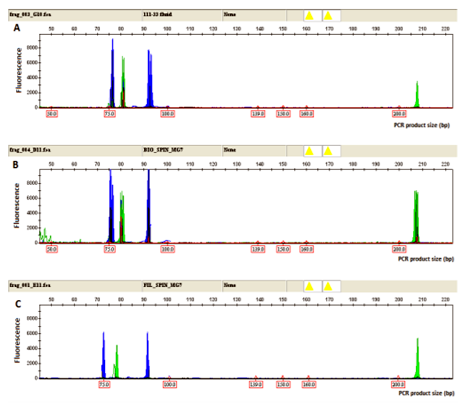
To prove the specificity and reproducibility of the extraction method from alternative materials, we simultaneously tested three types of materials derived from the same museum specimens: tissue, filter paper, and conservation fluid. The results showed the same, replicable PCR profile of ABO and amelogenin genotyping in all three types of material from the same museum specimen. The same, replicable PCR profile was obtained analyzing all sets of museum specimens, excluding one sample that yielded negative results for all three types of material (Figure 3, Table 4).
4. Discussion
This article demonstrates a non-destructive method of DNA extraction from museum specimens stored in liquid media. In the present study we successfully obtained DNA from museum specimens (tissue) and related materials (filter paper and conservation fluid) of sufficient quantity and quality for PCR despite their partial degradation.
We decided to use the ABO blood group systems as a test method for two reasons. First, this assay has many amplicons with suitable sizes. Second, the method is rather difficult and, in our opinion, good results constitute an optimal prognostic factor for other applications, including NGS techniques. The amelogenin primers in the PCR assay were applied to test the ability to amplify longer fragments.
Degradation of genetic material is the main problem that needs to be overcome when working with historical samples. A large body of literature reports a number of different approaches to DNA isolation from museum specimens in order to increase the yield and quality of the obtained genetic material. A number of conditions, including the age of the museum specimens as well as that of the preservation agent, are crucial for the quality of obtained DNA (Paireder 2013; Niland 2012; Hofreiter 2012). In present study we successfully obtained DNA from 100-year-old museum samples; this DNA subsequently worked well in a PCR assay, with a very high level of reliability. The negative aspect of the results described here is the relatively high level of degradation DNA extracted from conservation fluid as full profiles were obtained in only 25% of cases. However, short amplicons up to 100 bp were successfully amplified in more than 60% of cases.
One issue that might interfere with this method is chemical contamination (mainly by ethanol and glycerol) of DNA from a conservation agent. All three DNA purification methods used in this study ended with purified DNA. Applying the column-based DNA isolation method enabled us to obtain ethanol- and glycerol-free DNA from tissue and filter paper. Furthermore, use of a 5% Chelex-based method, which relied on heating of the sample to 98°C, purified the DNA via the evaporation of ethanol and other contaminants. The conservation fluid was purified by dialysis of the ethanol and glycerol from the solution containing the DNA. Extracted DNA was successfully purified, as proved by positive PCR results.
Museum specimens are often unique and very precious. Classical DNA isolation methods damage museum materials during the initial step due to tissue digestion. Therefore, alternative, non-destructive methods have been developed by numerous investigators (Rohland 2004; Bolnik 2012; Rohland 2007; Hervella 2012; Hofreiter 2012). In this study we demonstrated that materials related to museum specimens, such as the filter paper used for filtration of the conservation fluid and the conservation fluid itself, may be excellent alternative sources of genetic material. A similar approach was adopted by Shokralla et al (2010) who also showed that DNA could be extracted from conservation fluid, however, these authors proposed a different method of DNA extraction and purification. It must be pointed out that our museum samples were much older than Shokralla’s samples, which were only 10 years old). Furthermore, in that study positive results were obtained for short fragments (approximately 130 pb), but none at all for longer fragments. In addition to conservation fluid we used filter paper, which yielded better results in that we were able to amplify longer fragments (amelogenin was detected in almost 90% of all samples compared to 25% positive results for all dialyzed conservation fluids). It is worth emphasizing that DNA extracted from filter paper used for conservation fluid filtration yielded the same results as DNA extracted directly from tissue with a rate of success of approximately 90%. What is more, positive results were obtained even for longer fragments i.e. over 200 bp. One sample failed in PCR, but the real reason for this was a bullet present in the sample (a spinal cord), in this case the negative results were obtained for both tissue and filter paper. Lead and other metals from the bullet inhibited polymerase in PCR.
5. Conclusion
In summary, we proved that DNA could be successfully extracted from materials related to museum specimens such as filter paper used for conservation fluid filtration or conservation fluid itself. DNA extracted in this way yielded the same, reliable results as results obtained from the tissue. DNA extracted from the filter paper seemed to work better in PCR and demonstrated a lower rate of degradation. In the authors opinion, this material might be an excellent source of DNA suitable for PCR, but the question of whether such DNA can be used in other applications including NGS, still needs to be answered.
It is necessary to point out that this method can be applied only to museum specimens stored in liquid media; however, it is the first method to enable the preservation of unique tissues concurrently with DNA extraction.
References
- Bi, Ke, Linderoth Tyler, Vanderpool Dan, Good Jeffrey M, Nielsen Rasmus, and Moritz Craig. 2013. Unlocking the vault: next generation museum population genomics. Molecular Ecology 22:6018–6032.
- Bolnik, Deborah A, Bonine Holly M, Mata-Miguez Jaime, Kemp Brian M, Snow Meradeth H, and LeBlanc Steven A. 2012. Nondestructive Sampling of human skeletal remains yields ancient nuclear and mitochondrial DNA. Am J Phys Anthropol 147:293-300.
- Bonin, Serena, Hlubek Falk, Benhattar Jean, Denkert Carsten, Dietel Manfred, Fernandez Pedro L, Höfler Gerald, Kothmaier Hannelore et al. 2010. Multicentre validation study of nucleic acids extraction from FFPE tissues. Virchows Arch 475:309-17.
- Gilbert, M Thomas P, Moore Wendy, Melchior Linea, and Worobey Michael. 2007. DNA extraction from dry museum beetles without conferring external morhplogical damage. PLoSOne 3:772-776
- Hervella, Montserrat, Iňiguez Maitane G, Izagirre Neskuts. Anta Alberto, and de-la-Rúa Concepción. 2012. Nondestructive methods for recobery of biological material from human teeth for DNA extraction. J Forensic Sci 60:136-141.
- Hofreiter, Michael. 2012. Nondestructive DNA extraction from museum specimens. Methods Mol Biol 840:93-100.
- Jiang, Xianhua, He Juan, Jia Fei, Shen Hongying, Zhao Jinling, Chen Chuguang, Bai Liping, Liu Feng, et al. 2012. An integrated system of ABO typing and multiplex STR testing for forensic DNA analysis. Forensic Science International: Genetics 6:785-797.
- Niland, Erin N, McGuire Audrey, Cox Mary H and Sandusky George E. 2012. High quality DNA obtained with an automated DNA extraction method with 70+ year old formalin-fixed celloidin-embedded (FFCE) blocks from the Indiana Medical History Museum, Am. J. Transl 4:198–205.
- Paireder, Stefan, Werner Bettina, Bailer Josef, Werther Wolfgang, Schmid Erich, Patzak Beatrix, and Cichna-Markl Margit. 2013. Comparison of protocols for DNA extraction from long-term preserved formalin fixed tissues. Anal Biochem 439:152-160.
- Rohland Nadin, Siedel Heike, and Hofreiter Michael. 2004. Nondestructive DNA extraction method for mitochondrial DNA analysis of museum specimens. BioTechniques 36:814-821.
- Rohland Nadin, and Hofreiter Michael. 2007. Ancient DNA extraction from bones and teeth. Nat Protoc 2:1756-1762.
- Shokralla, Shadi, Singer Gregory AC, Hajibabaei Mehrdad. 2010. Direct PCR amplification and sequencing of specimens’ DNA from preservative ethanol. BioTechiques 44:233-234
- Thomsen, Philip Francis, Elias Scott, Gilbert M Thomas, Haile James, Munch Kasper, Kuzmina Svetlana, Froese Duane G, Sher Andrei, et al. 2009. Non-destructive sampling of ancient insect DNA. PloSOne 4:5048-5054
- Tin, Mandy Man Ying, Economo Evan Philip, and Mikheyev Alexander. 2014. Sequencing degraded DNA from non-destructively sampled museum specimens for RAD-tagging and low-coverage shotgun phylogenetics. PLoSOne 9:96793-96802
- Wandeler, Peter, Hoeck Paquita E A, and Keller Lukas F. 2007. Back to the future: museum specimens in population genetics. Trends in Ecology and Evolution 22:634–642.
- Wong Wing Hing, Tay Ywee Chieh, Puniamoorthy Jayanthi Balke Michael, Cranston PS, Meier R. 2014. Direct PCR optimization yields a rapid, cost-effective, nondestructive and efficient method for obtaining DNA barcodes without DNA extraction. Mol Ecol Resour 14:1271-1280.
- Yang Hong, Golenberg Edward M, and Shoshani Jeheskel. 1997. Proboscidean DNA from museum and fossil specimens: an assessment of ancient DNA extraction and amplification techniques. Biochem Genet 35:165-79.