


|
|
DOI Prefix 10.20431 |
Information
Journal Policies
Turcot Syndrome with Gliosarcoma: A rare variation of a Lynch Syndrome Phenotype
Laura Baugh,Michelle Shiller
Copyright :© 2017 Authors. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Familial cancer syndromes are unique in that genetic testing can facilitate determining which affected relatives should be recommended for early surveillance, the results of which may have a profound effect. More than 50 familial cancer syndromes have been described, one of which is Lynch syndrome. Lynch syndrome involves the mutation of specific proteins that function in DNA mismatch repair. A phenotypic variant of Lynch syndrome, Turcot syndrome, has been defined as the presence of synchronous or metachronous colorectal and primary central nervous system (CNS) tumors, historically glioblastoma multiforme, in the presence of a documented mutation in one of the mismatch repair (MMR) proteins. Here we report the case of a 34 year old male who initially presented with altered mental status and seizures. Clinically, he had synchronous sigmoid colon and rectal tumors as well as gliosarcoma, an unusual, higher-grade variant of glioblastoma multiforme. Tumor testing confirmed loss of MMR protein expression in both gastrointestinal and CNS lesions, microsatellite instability by PCR, and a germline mutation in MSH2 was detected in peripheral blood.
Keywords: Turcot syndrome, Lynch syndrome, gliosarcoma, mismatch repair,Clinical Case Reports
1.Introduction
In the post-genomic era of medicine, the association of specific mutations in familial cancer syndromes and their association with specific tumor types and/or histologic variants are actively being identified and documented. Newly detected alterations increase our understanding of these syndromes, and each patient identified improves our awareness of the varying phenotypes and genotype-phenotype correlations. Integration of clinical findings with new technologies that identify genetic findings in the patient’s tumor as well as germline DNA, together with additional clinical tests, continues to facilitate an approach to patients that is more informed and effective, including opportunities for genetic counseling to further educate and empower patients and their families. The case presented herein illustrates this through identification of a patient with type I Turcot syndrome, a Lynch syndrome variant, with confirmatory germline genetic testing and establishment of the molecular pathophysiologic explanation (i.e, microsatellite instability) for his disease.
Specifically, this case was unique in that the patient’s CNS tumor consisted of an unusual histologic subtype, of which only two other cases have been documented. Our goal is to contribute to the rapidly growing cohort of individuals with Lynch Syndrome, including unique subsets, in order to better understand clinical associations and facilitate optimal surveillance of affected relatives.
2. Case
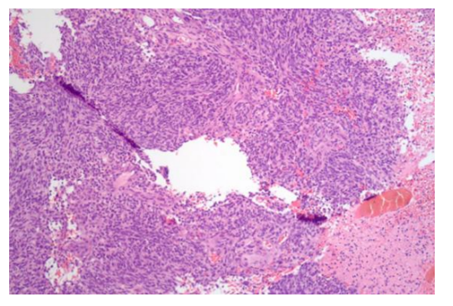
In February 2014, a 34 year old African American male was admitted to Baylor Medical Center Carrollton with altered mental status and a recent history of generalized seizures while driving. The patient had an unremarkable medical history; his family history was notable for a maternal aunt with pancreatic cancer. An MRI scan showed a complex heterogeneous mass within the right temporal lobe measuring approximately 3.5 x 3.1 x 2.9 cm with a mild mass effect, favored to be a primary brain malignancy. Subsequent follow up with neurosurgery including a pre-operative MRI performed a month later at Baylor Dallas showed the right temporal lobe mass to have increased in size to 6.8 x 5.3 x 4.3 cm with interval development of subfalcine and uncal herniations and hydrocephalus. He underwent right temporal parietal craniotomy on March 18, 2014 with tumor resection. Intraoperative consultation provided a diagnosis of high-grade glioma. The post-operative MRI one day later showed a small residual enhancing component along the posterior surgical margin. Following fixation, the expected features of glioblastoma multiforme were present. In addition, pleomorphic, round to spindled cells with extensive spindled cells in the background and a fibrotic stroma permitted a more specific diagnosis of gliosarcoma on permanent sectioning (Figure 1). Immunoperoxidase stains were performed and positive for Glial Fibrillary Acidic Protein and p53, with a very high proliferative index (approximately 80%) by MiB-1, therefore WHO grade IV. The patient was discharged several days after the operation and was started on temozolomide; radiation was also performed.
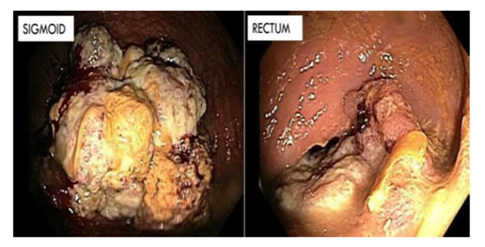
A CT scan of the abdomen was performed which incidentally showed a 6.5 cm fungating sigmoid mass. Synchronous colorectal primary tumors were consequently identified on colonoscopy as follows: 1) 7 cm firm, fixed non-circumferential rectal mass, 6 to 7 cm from the anal verge and 2) a completely obstructing circumferential mass in the sigmoid colon. (Figure2). Biopsies demonstrated moderately differentiated adenocarcinomas arising in polyps in both locations. Given this, the patient underwent genetic counseling, testing was ordered, and the patient’s family was recommended to also undergo genetic testing, assuming a clinical diagnosis of Lynch Syndrome. Next generation sequencing of the patient’s blood of EPCAM, MSH2, MSH6, and MYH identified a deleterious single base deletion (867delT) in the MSH2 gene, confirming Lynch Syndrome. In July 2014 the patient underwent abdominoperineal resection (APR) for synchronous carcinomas of the rectum and sigmoid colon.
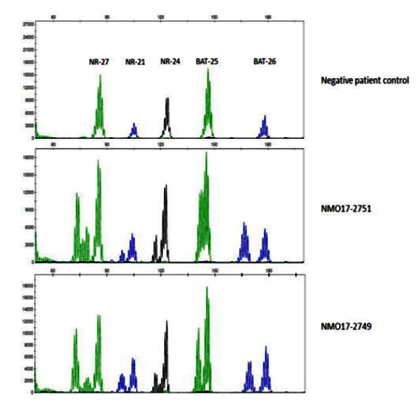
Gross examination of the APR confirmed the synchronous primaries, the larger lesion located in the sigmoid colon. Grossly and microscopically the tumor invaded through the muscularis propria into the perirectal fat with negative margins (pT3). Metastatic disease was confirmed with five of fifty-six lymph nodes positive (pN2a), stage IIIB. Tumor infiltrating lymphocytes were present in the lesions. Immunohistochemistry for the mismatch repair (MMR) proteins demonstrated loss of nuclear MSH2 and MSH6 expression suggestive of Lynch syndrome, and microsatellite instability high was detected on the tumor by PCR, further supporting this possibility (Figure 3).
In December 2014, surveillance MRI revealed a mass along the inferior margin of the right temporal resection cavity with a new large region of enhancement involving the majority of the remaining right temporal lobe. In late December 2014 the patient had a right temporal craniotomy for resection of recurrent glial neoplasm and implantation of carmustine wafers. The tumor was diagnosed as residual high grade glioma with features of gliosarcoma and demonstrated a high MiB-1 labeling index exceeding 80%, with strong nuclear expression of p53 by immunohistochemistry, consistent with the original resection. Follow-up MRIs continued and in May 2015 identified multifocal enhancing lesions within the right temporal resection bed as well as separate right thalamic and left caudate lesions. Biopsy from the resection bed found fewer sarcomatous features indicating that the right temporal mass was evolving, with a diagnosis of glioblastoma for this biopsy. In June 2016 the patient was discharged to a rehabilitation facility.
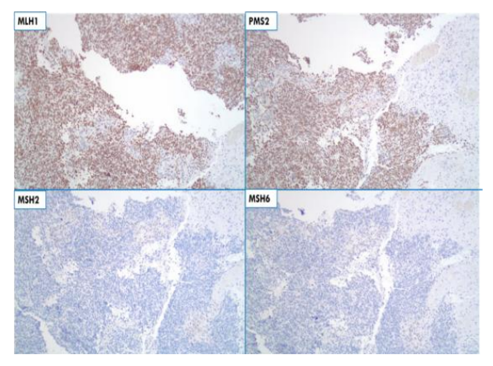
Subsequent tumor testing of the patient’s gliosarcoma by immune histo chemistry confirmed loss of nuclear expression for MSH2 and MSH6 (Figure 4). These results, in conjunction with the patient’s synchronous colorectal adenocarcinomas (with similar findings) and gliosarcoma, a rare central nervous system tumor variant, demonstrate an unusual case of Turcot syndrome.
3. Discussion
Lynch syndrome is an autosomal dominant hereditary cancer syndrome that is the most common hereditary form of colorectal and endometrial cancer. It has been estimated to be responsible for 1 to 6% of all colorectal cancer cases, formerly known as hereditary non-polyposis colorectal cancer syndrome (HNPCC) [1]. The past 20 years of experience with Lynch Syndrome has identified numerous extra-colonic manifestations including: endometrial, small intestine, ovarian, renal pelvis, pancreatic, gastric, liver and biliary, sebaceous, and CNS (historically glioblastoma multiforme) tumors. The incidence of these extra-colonic tumors varies, with endometrial the most prevalent. Brain tumors are one of the least common manifestations. Lynch syndrome is associated with the aberrant function of one of the DNA mismatch repair genes MSH2, MSH6, MLH1, or PMS2. Epigenetic interference with gene expression includes methylation of the MLH1 promoter or exon deletions of TACSTD1, the gene encoding EPCAM, which epigenetically inactivates MSH2 [2]. Such modifications are important to note, especially EPCAM, because in order for assessment of MSH2 mutations to be considered complete, EPCAM testing must be a component of that testing. A key indicator that such testing is needed is the information provided by functional testing with loss of expression of either MSH2 in isolation (which is rare), or concomitant loss of MSH2 and MSH6 (more common). The incidence of mutations in MSH2 have been disputed, some claiming greater prevalence of extra-colonic manifestations [3], and others not [4]. The case we present involves a deleterious deletion evidenced by loss of nuclear expression of MSH2 and MSH6, and an MSI-H phenotype. Alterations at base 867 of MSH2 occur in the helix-turn-helix domain of the gene, critical to its function.
Turcot, in 1959, first reported the case of two young siblings with diffuse polyposis of the colon that were subsequently found to have primary tumors of the central nervous system [5]. In this context, Turcot syndrome was a component of FAP. The eponym resulting from the original case report describing features of the syndrome was ultimately also applied to Lynch syndrome. In keeping with the original description, Turcot syndrome in individuals with germline mismatch repair deficiency is characterized by synchronous or metachronous colorectal and central nervous system tumors. In 1995 the molecular basis of the syndrome was more clearly defined by Hamilton et al who described a form with predominantly APC mutations resulting in characteristic brain tumors, predominantly childhood medulloblastoma [6]. The second form was found to harbor mutations in the mismatch repair genes and to predispose to central nervous system tumors, predominantly glioblastoma. Other tumor types involved include astrocytoma, neuroblastoma, and gliosarcoma [[7], [8]]. One low grade astrocytoma in Turcot was associated with IDH1 expression [9]. Turcot syndrome due to hereditary defects in mismatch repair protein function is relatively rare with approximately 150 cases reported in the literature [10].
Using a national cohort of Lynch syndrome families from Finland, Gylling et al found that brain tumors were diagnosed at the average age of 38 years and that glioma was the most common histologic subtype [11]. Watson et al utilized a cohort of 6,176 members of 261 families to document that the risk of brain cancer in Lynch syndrome showed an overall lifetime risk of 2% [12]. The predominant gene involved in Turcot syndrome was initially reported to be PMS2 which has since been revised and appears to be more highly associated with mutations in MSH2 [[8], [12], [13]. This hypothesis is further supported and provides additional evidence for a genotype phenotype correlation with individuals who have germline MSH2 mutations and Turcot syndrome.
Regarding the histology, the most common type of brain tumor associated with Turcot is glioblastoma which is a high grade astrocytoma with nuclear atypia, cellular pleomorphism, and mitotic activity and concomitant necrosis or microvascular proliferation. Interestingly, there have been two other documented cases of gliosarcomas in Turcot syndrome: one in a 24 year old female and the other in a 32 year old male in a Danish Lynch syndrome cohort [[8], [14]], both with mutations in MSH2. We here report a third case of Turcot syndrome in a 34 year old man with gliosarcoma and synchronous colorectal neoplasms, also with a mutation in MSH2. The genotypic abnormalities in the first two documented cases consisted of a MSH2 point mutation (c.942 + 3A>T) in the female and an MSH2 insertion and substitution mutation (c.1696_1697AA>G) in the male. The patient we present here had an MSH2 mutation (c.867delT) with loss of expression of both MSH2 and MSH6 and MSI-H phenotype. The histology of his initial tumor resection was negative for IDH1 expression and positive for diffuse p53 expression. All three of these mutations are predicted to be deleterious for the expression and function of MSH2.
More recently, the FDA made a landmark decision by approving a drug indication based on a molecular finding. Specifically, any tumor that has deficient mismatch repair protein expression or microsatellite instability is a candidate for immune therapy targeted at the PD-1 receptor. Targeting the PD-1 receptor re-invigorates the innate immune response, permitting the local host response to have anti-tumor effect [15].
4. Conclusion
The importance of diagnosing Lynch syndrome, or any hereditary cancer syndrome, early is a topic of worthwhile emphasis. Advantages of early detection include the implementation of prevention strategies including specialized cancer screening, heightened surveillance, and prophylactic surgeries [16]. Moreover, detecting cancer early, or preventing it, also decreases healthcare cost not only with managing a diagnosed malignancy but also with treating comorbidities that may or may not arise due to treatment. Additionally, knowing whether or not the patient has Lynch Syndrome helps guide therapy in a similar vein as knowing the BRCA1/BRCA2 status in breast cancer. Individuals with Lynch Syndrome and colorectal carcinoma show a decreased response to 5-fluorouracil, and the surgeon may change their surgical management based on this knowledge [17].
Almost 60 years after Turcot described the “potentialities” of the mutant gene associated with his initial description of Turcot syndrome, we have a much better understanding of the subtypes of Turcot syndrome, including the spectrum of genes involved and the variety of “potentialities,” or additional tumors that can evolve with Lynch syndrome. With the advent of next generation sequencing and the ability to more widely interrogate genes, polymorphisms and variants of uncertain significance in the mismatch repair genes are being detected with greater frequency. Careful record-keeping of the incidence of these less understood alterations, together with family studies, provides hope that better correlation of genotype with phenotype will be of clinical value in determining the significance of such genetic aberrations [18],[19].
Acknowledgments
We would like to acknowledge Dr. Sarah Boostrom for figure 2, endoscopic images of the rectosigmoid tumors and Dr. Michael Weindel for figure 3, the microsatellite stability signatures.
References
- Lynch H, Smyrk T, Watson P, Lanspa S, Lynch J, Lynch P, Cavalieri R, Boland R. Genetics, natural history, tumor spectrum, and pathology of hereditary nonpolyposis colorectal cancer: an updated review. Gastroenterology 1993; 104(5):1535-1549.
- Ligtenburg M, Kuiper R, Chan T, Goossens M, Hebeda K, Voorendt T, Lee T, Bodmer D, Hoenselaar E, Hendriks-Cornelissen S, Tsui W, Kong C, Brunner H, van Kessel A, Yuen S, van Krieken J, Leung S, Hoogerbrugge N.Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3’ exons of TACSTD1. Nat Genet 2009;41(1): 112-117.
- Geary J, Sasieni P, Houlston R, Izatt L, Eeles R, Payne S, Fisher S, Hodgson S. Gene-related cancer spectrum in families with hereditary non-polyposis colorectal cancer. Familial Cancer 2008;7:163-172
- Vasen H, Stormoken A, Menko F, Nagengast F, Kleibeuker J, Griffioen G, Taal B, Moller P, Wijnen J. MSH2 mutation carriers are at higher risk of cancer than MLH1 mutation carriers: a study of hereditary nonpolyposis colorectal cancer families. J Clin Oncol 2001; 19(20):4074-4080.
- Turcot J, Despres J, Pierre F. Malignant Tumors of the Central Nervous System Associated with Familial Polyposis of the Colon: Report of Two Cases. Dis Colon Rectum 1959; 2:465-468.
- Hamilton S, Liu B, Parsons R, Papadopoulos N, Jen J, Powell S, Krush A, Berk T, Cohen Z, Tetu B, Burger P, Wood P, Taqi F, Booker S, Petersen G, Offerhaus G, Tersmette A, Giardiello F, Vogelstein B, Kinzler K. The molecular basis of Turcot’s syndrome. N Engl J Med 1995;332(13): 839-847
- Baehring J, Hui O, Piepmeier J, Bannykh S. Anaplastic oligoastromcytoma in Turcot syndrome. J. Neurooncol 2009, 95:293-298.
- Therkildsen C, Ladelund S, Rambech E, Persson A, Petersen A, Nilbert M. Gliobla stomas, astrocytomas and oligodendro gliomas linked to Lynch syndrome. Eur J Neurol 2015; 22:717-724.
- Alkhotani A, Ambus I, Velsher L, Rowsell C, Keith J. IDH1 mutated low grade astrocytoma occurring in MSH2 mutated Lynch syndrome family. Hum Pathol: Case Reports 2016; 6:45-47.
- Ozerov S, Zakharov I, Talypov S, Konovalov D, Kislyakov A, KachanovD, Zheludkova O, Varfolomeeva S, Rachkov V. Turcot syndrome. A rare case and literature review. J Neurosurg 2013; 3:46-49.
- Gylling A, Nieminen T, Abdel-Rahman W, Nuorva K, Juhola M, Joensuu E, Jarvinen H, Mecklin J, Aarnio M, Peltomaki P. Differential cancer predisposition in Lynch syndrome: insights from molecular analysis of brain and urinary tract tumors. Carcinogenesis 2008; 29(7):1351-1359.
- Watson P, Vasen H, Mecklin J, Bernstein I, Aarnio M, Jarvinen H, Myrhoj T, Sunde L, Wijnen J, Lynch H. The risk of extra-colonic and extra-abdominal cancer in the Lynch syndrome. Int J Cancer 2008; 123:444-449.
- Lucci-Cordisco E, Zito I, Gensini F, Genuardi M. Hereditary nonpolyposis colorectal cancer and related conditions. Am J M Genet 2003; 122(A):325-334.
- Nilbert M, Therkildsen C, Nissen A, Akerman M, Bernstein I. Sarcoma associated with hereditary nonpolyposis colorectal cancer: broad anatomical and morphological spectrum. Familial Cancer 2009; 209-213.
- Le D., Uram J, Wang H, Bartlett B, Kemberling H, Eyring A, Skora A, Luber B, Azad N, Laheru D, Biedrzycki B, Donehower R, Zaheer A, Fisher G, Crocenzi T, Lee J, Duffy S, Goldberg R, de la Chapelle A, Koshiji M, Bhaijee F, Huebner T, Hruban R, Wood L, Cuka N, Pardoll D, Papadopoulos N, Kinzler K, Zhou S, Cornish T, Taube J, Anders R, Eshleman J, Vogelstein B, and Diaz, Jr. L. PD-1 blockade in tumors with mismatch-repair deficiency. NEJM 2015; 372:2509-2520.
- Kastrinos K, Stoffel E. History, genetics, and strategies for cancer prevention in Lynch syndrome. Perspectives in Clinical Gastro-enterology and Hepatology 2014; 12:715-727.
- Kawakami H, Zaanan A, Sinicrope F. MSI testing and its role in the management of colorectal cancer. Curr Treat Options Oncol. 2015; 16(7): 30.
- Buerstedde J, Alday P, Torhorst J, Weber W, Muller H, Scott R. Detection of new mutations in six out of 10 Swiss HNPCC families by genomic sequencing of the hMSH2 and hMLH1 genes. J Med Genet 1995; 32:909-912.
- Martin-Lopez J, Barrios Y, Medina-Arana V, Andiyar M, Lee S, Gu L, Li G, Ruschoff J, Saledo E, Fishel R. The hMSH2 (M688R) Lynch syndrome mutation may function as a dominant negative. Carcinogenesis 2012; 33(9):1647-1654