


|
|
DOI Prefix 10.20431 |
Information
Journal Policies
Holt Oram Syndrome-Revisited: Clinical Images
Dr.Shivani Rao1,Dr.P.C.Negi2,Dr.KunalMahajan3,Dr.Saurabh Galodha4
2.Professor Department of Cardiology, Indira Gandhi Medical College, Shiml , HP, India.
3.Senior Resident Department of Cardiology, Indira Gandhi Medical College, Shimla, H, India.
4.Assistant Professor Department of Surgery, Indira Gandhi Medical College, Shimla, HP, India.
Copyright : © 2017 Authors. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
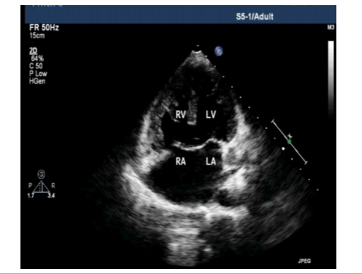
A 24-year male presented with history of central cyanosis since early childhood. Patient denied any history of any recurrent respiratory tract infections and delayed growth milestones. On examination there was right upper limb abnormality in the form of foreshortened arm, underdeveloped small sized thumb and there was difficulty in apposition of thumb to fingertips. Metacarpals and right radius was underdeveloped. (Figure.1 and 2) ECG revealed left axis deviation, broad notched P wave with biventricular hypertrophy. Chest X-ray showed a normal sized heart with decreased pulmonary vascularity and a concave pulmonary arterial segment. Echocardiography showed complete atrio-ventricular septal defect (AVSD) (Figure. 3) with pulmonary stenosis (PS). A complete Diagnosis of Holt Oram syndrome (HOS) with AVSD with PS was made. Patient underwent successful intracardiac repair.
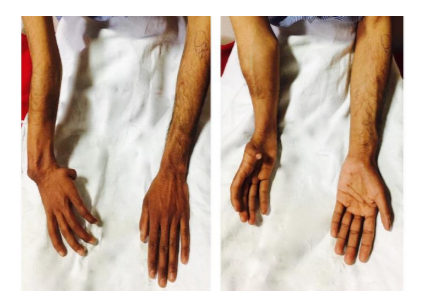
Holt oram syndrome was first described in 1960 [1] and is also known as the atriodigital dysplasia syndrome It is a rare autosomal dominant genetic disorder .Several mutations have been described, but the most frequent is in the TBX5 gene of the T-box complex, located on chromosome 12q24.1 [2]. It is clinically characterized by congenital cardiac defects and morphological abnormalities of the upper limbs. Defective development of the embryonic radial axis (e.g. hypoplasia, aplasia, fusion, or other anomalous development) results in a wide spectrum of phenotypes and may manifest as thumb being absent, underdeveloped or triphalangeal.[3] There may be malformations of the metacarpals, hypoplastic or absent radii, ulna or humerus and scapulae may be absent or abnormal. The above mentioned abnormalities may be unilateral or bilateral and asymmetric. Majority of patients also have cardiac malformations and almost every type of cardiac anomaly has been reported, either singly or as part of a group of multiple defects. The most common are atrial septal defects mostly secundum type, ventricular septal defects, abnormal isomerism and anomalous pulmonary venous return. Rare but other cardiac associations include pulmonary stenosis, mitral valve prolapse, and arrhythmias in the form of atrioventricular blocks. More complex cardiac lesions such as tetralogy of Fallot, AVSD, and total anomalous pulmonary venous return are also noted in subjects with HOS [4].
References
- Holt M, Oram S. Familiar heart disease with skeletal malformation. Br Heart J 1960;22:236:42.
- C. T. Basson, D. R. Bachinsky, R. C. Lin et al., “Mutations in humans cause limb and cardiac malformations in Holt-Oram syndrome,” American Journal of Medical Genetics, vol. 65, pp.128–132, 1996.
- G. Oður, D. Gül, M. K. Lenk, N. Imirzalioðlu, F. Alpay, and E. Oður, “Variable clinical expression of Holt-Oram syndrome in three generations,” The Turkish Journal of Pediatrics, vol.40, pp. 613–618, 1998.
- L. J. Sletten and M. E. Pierpont, “Variation in severity of cardiac disease in Holt-oram syndrome,” American Journal of Medical Genetics, vol. 65, pp. 128–132, 1996.