


|
|
DOI Prefix 10.20431 |
Information
Journal Policies
Analysis of the Complete Genome Sequence Reveals a High Degree of Conservation of the Hiv-1 Subtype B in Cuba
Madeline Blanco de Armas1,Liuber Yans Machado Zaldivar2,Hector M.Diaz Torres3,Enrique Noa4,Dania Mercedes Romay Franchi5
2.Hospital Hermanos Ameijeiras, Havana, Cuba.
Copyright : © 2017 . This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The analysis of the structural genes of HIV-1 in different Cuban patients showed a preserved behavior for subtype B, which motivated to study the complete length of the viral genome in patients with this pattern and to demonstrate the high degree of conservation of this genetic variant. More than 8,000 bp of the HIV-1 genome were sequenced from four HIV-positive individuals with no epidemiological linkage, previously identified as subtype B by pol gene analysis. Phylogenetic and recombination analysis showed the conserved pattern of circulating subtype B in the Cuban seropositive population. The results of the present study strengthened the epidemiological surveillance system of the National STI / HIV / AIDS Program in Cuba.
HIV-1, full genomes, molecular epidemiology, subtype B, Cuba,AIDS
1. Introduction
The complete HIV-1 genome sequence was first published in 1985 by Ratner et al[1] and subsequently numerous reports documented the extraordinary genetic diversity of HIV-1, derived from the high rate of mutations and recombination of this virus [2]. Frequency of circulating recombinant forms (CRFs) and unique recombinant forms (URFs) has increased in multiple geographic areas, where they co-circulate different genetic forms [3]. Population movements have also led to the spread and epidemic expansion by the world of genetic variants and recombinant forms [4,5]. On the other hand, the event of transmission of the same genetic form and its propagation within a group with specific risk behaviors (founding effect), has resulted in the establishment of the epidemic in that area, where certain genetic variants predominate, over others. However, due to the rapid evolution of HIV and its global spread, other variants emerge and recombine [6,7].
In Cuba, in the 1980s, subtype B had its founding effect on the population of men who have sex with men (MSM), which is also the majority risk group [8]. Other non-B subtypes were introduced by people heterosexual men, who acquired the infection in different African countries and, on their return, transmitted it to their (female) sexual partners and have spread less in the HIV-positive population [9,10]. After more than two decades, the progressive appearance of different CRFs and multiple URFs has been observed, due, among other causes, to the recombination of the subtypes circulating in Cuba . Different studies carried out in Cuba for the molecular characterization of the structural genes of HIV-1 showed in patients characterized as subtype B, a conserved pattern, for each of the genes studied [11-16]. Subtype B is the genetic variant of HIV-1 most disseminated in the Cuban epidemic from its beginnings to the present day, so it is of interest to study the complete genome length in patients with this pattern and to demonstrate the high degree of conservation observed in the predominant subtype in Cuba.
2. Materials And Methods
Ten mL of whole blood were collected from four HIV-1 seropositive patients with no documented epidemiological link and previously classified as subtype B according to the pol gene. The proviral DNA was obtained using the High Pure Viral Nucleic Acid Kit (Roche Diagnostics GmbH, Germany) according to the manufacturer's instructions.
The viral load, at the time of sampling, was determined using the Cobas AmpliPrep - Cobas TaqMan HIV-1 test version 2.0 and the CAP-CTM 48 (Roche) kit. The CD4 values in percent and absolute value at the time of sampling were obtained from the SIDATRAT database of the Institute of Tropical Medicine Pedro Kouri.[17]
Sequencing of the complete HIV-1 genome was performed according to the protocol described by Nadai et al in 2008[18] and using the CEQ Dye-labeled dideoxy terminator cycle sequen - cing kit (Beckman Coulter, Company. Beverly, United States United).
The sequences were assembled and edited using the Sequencher v 5.0 program (GenCode Corporation), for further realization of a BLAST (http://www.ncbi.nlm.nih.gov/BLAST). The subtype was determined by the REGA HIV-1 sub typing tool. (http: //bioafrica.mrc. Rega-genotype-3.0.2 / hiv / typin gool).
Multiple sequence alignment was performed through the Muscle program present in the MEGA v 6.0 [19]. Samples were aligned with subtype B sequences from the Americas, Europe, Asia and Africa obtained from the HIV database -1 of Los Alamos (Supplementary material 1). The analysis for the determination of possible recombinants was performed using the RDP3 program.
For the selection of the best nucleotide substitution model, the jModel Test v 2.1.4 program was used. Phylogenetic trees were constructed using the maximum likelihood method, using the GTR + G + I model [20]. To verify the support of the branches and calculate the numbers near the nodes, the SH-like and boostrap tests were used (1000 Replicates) and considered reliable, those with values greater than 0.90 and above 70% [21,22]. The PhyML v 3.0 was used. The trees were visualized and edited by the Fig Tree program. Mutations associated with antiretroviral resistance were determined according to the algorithm available in the Stanford Database (http://hivdb.stan ford.edu). The prediction of viral tropism was determined using the geno2pheno coreceptor tools (http // www.coreceptor.geno2pheno.org) [23] and WebPSSM (https://indra.mullins.micr obiol. Was hington.edu/webpssm/) [24].
3. Results And Discussion
The four patients included in the study were male and resided in Havana and had no epidemiological link. The sexual orientation of the patients studied was MSM. Table 1 illustrates the epidemiological and clinical characteristics of the patients analyzed.Splicing of approximately 8000 bp was achieved, with the exception of the LTR sequences. The analyzed samples were classified as subtype B by the REGA HIV sub typing tool program and no presence of recombination was detected by analyzing the genome of the patients studied, as illustrated in the analysis of sample 12CU087 (Figure 1)
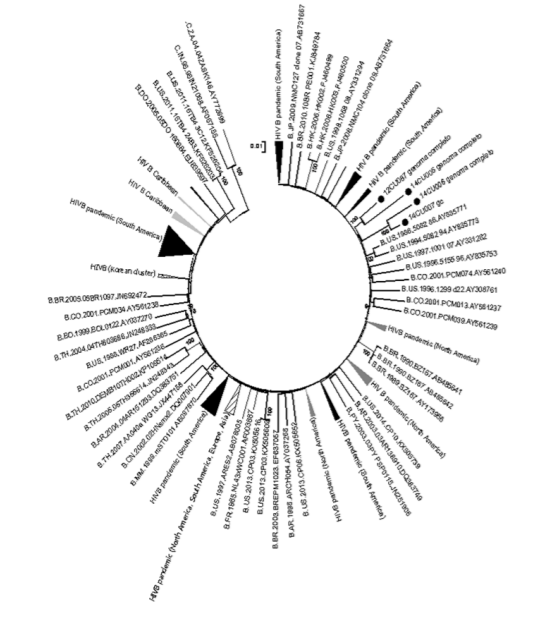
Phylogenetic analysis grouped the Cuban sequences with sequences belonging to the pandemic subtype B (Figure 2). Samples 12CU087 and 14CU05 were grouped with sequences from the United States and France, while samples 14CU06 and 14CU07 were related to sequences from Colombia, South America.<
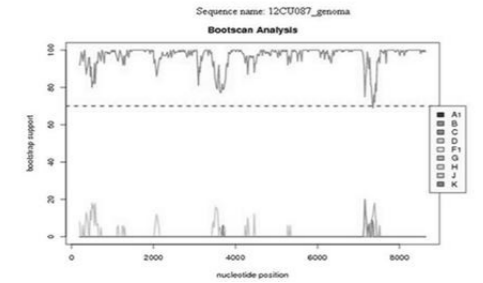
The HIV / AIDS epidemic in Cuba has been characterized since its beginnings to present a high genetic diversity, translated in the wide variety of introduced subtypes and the circulation of recombinant forms [9-16]. The first molecular epidemiology studies of HIV-1 in Cuba were directed to the most hyper variable region of the HIV-1 env gene and showed a predominance of subtype B and association of this variant in the population of MSM . The use of highly active antiretroviral therapy (HAART) in Cuba since 2001 and the subsequent introduction of genotypic assays for the determination of HIV-1 resistance to antiretroviral (ARV) allowed the analysis of a larger number of samples from treated and untreated patients. Although the results of the studies, mainly focused on the regions coding for the protease and reverse transcriptase enzymes of the pol gene, indicated an increase in recombinant forms, subtype B continued as a majority genetic variant in the Cuban seropositive population [15,16,25]. The high genetic diversity of HIV-1 described in Cuba favored the origin of new viral variants, to the detriment of pure subtypes. However, the founding effect of subtype B in the epidemiological context of our country and the origin of many of the sequences of this subtype as of 1977[26] can explain the conservation of the genome of this genetic variant in Cuba. Several studies have linked genetic diversity with the presence of mutations associated with HIV-1 resistance to ARV, with no relationship between these variables [27,28]. Non-detection of viral variants with ARV resistance profile favors the use of an effective HAART, which entailed the therapeutic success achieved by the patient identified with the sample number 12CU087. However, the presence of X4 and dual R5X4 viruses in two recently diagnosed patients is interesting. HIV-1 variants with these characteristics are most frequently detected in the final stages of infection, relating them to the rapid progression to AIDS [29] and the early approach to HIV diagnosis in high-risk and early onset populations of HAART, should be one of the main strategies of health systems in the world, if it is to stop the epidemic by 2020.
Subtype B had multiple introductions in Cuba and is phylogenetically related to sequences belonging to the pandemic variant that circulates mainly in North America and Europe 26, which supports the epidemiological history reported by various authors since the beginning of the HIV / AIDS epidemic in our country [9,10]. A recent study by Machado et al [26]presents the relationship of Cuban sequences with those from the United States, Canada and Europe. However, Junqueria et al., 2011, supports the origin of Cuban subtype B with South America [30]. Population movement and continuous exchange explain the phylogenetic relationship of the sequences studied with countries of North America, Europe and South America, respectively.
4. Conclusions
The above mentioned elements show a conserved pattern of subtype B in the samples studied, a result that feedback to the national STI / HIV / AIDS program in Cuba on the evolution of the majority genetic variant and its possible implications in the diagnosis, vaccine development And new therapeutic strategies.
Accession Number of Nucleotide Sequences
The accession numbers in the GenBank sequences studied in the present study are: KR914675-KR914678.
References
- Ratner L, Haseltine W, Patarca R, Livak KJ, Starcich B, et al. Complete nucleotide sequence 11of the AIDS virus, HTLV-III. Nature 1985; 313: 277–284.
- Rhodes T, Wargo H, and Hu W-S: High rates of human immunodeficiency virus type 1 recombination: Near-random segregation of markers one kilobase apart in one round of viral replication. J Virol 2003; 77:11193–11200.
- Thomson MM, Pérez Álvarez L, and Nájera R: Molecular epidemiology of HIV-1 genetic forms and its significance for vaccine development and therapy. Lancet Infect Dis 2002; 2:461–471.
- Osseo-Asare AD (2007). The African Aids Epidemic: A History. Social History of Medicine. 2007; 20: 401–402.
- Gilbert MT, Rambaut A, Wlasiuk G, Spira TJ, Pitchenik AE, et al. The emergence of HIV/AIDS in the Americas and beyond. Proc Natl Acad Sci USA. 2007; 104: 18566–70.
- Hue´ S, Pillay D, Clewley JP, Pybus OG. Genetic analysis reveals the complex structure of HIV-1 transmission within defined risk groups. Proc Natl Acad Sci USA. 2005; 102:4425–9.
- Perrin L, Kaiser L, Yerly S. Travel and the spread of HIV-1 genetic variants. The Lancet. 2003; 3: 22–27.
- Gorry C. Cuba’s National HIV/AIDS Program. MEDICC Review. 2011; 13(4): 5-8.
- Rolo F, Miranda L, Wainberg M, Gu Z, Lobaina L, Noa E. Envelope V3 Region Sequences of Cuban HIV-1 Isolates. J. Acquir Immune Defic Syndr 1995; 9:123-5.
- Gómez C, Iglesias E, Perdomo W, Rolo F, Blanco M, Duarte C. Isolates from four different HIV type 1 clades circulating in Cuba. Identified by DNA sequence of the C2-V3 region. AIDS Res Hum Retroviruses 2001; 17(1):55-8.
- Blanco M, Martínez N, Díaz HM, Lubián AL, Romay D, Regalado L. Detección de nuevos subtipos y formas recombinantes del VIH-1 en Cuba mediante el ensayo de movilidad del heteroduplex env / gag. Rev. Latinoam. Microbiología. 2002; 44 (4), Mx ISSN-0034-9771.
- Cuevas MT, Ruibal I, Villahermosa ML, Díaz H, Delgado E, Parga EV, et al. High HIV-1 genetic diversity in Cuba. AIDS 2002, 16:1643-1653
- Pérez L, Thomson M, Bleda MJ. HIV Type 1 Molecular Epidemiology in Cuba: High Genetic Diversity, Frequent Mosaicism, and Recent Expansion of BG Intersubtype Recombinant Forms. AIDS Res Hum Retroviruses 2006; 22 (8):724-33.
- Resik S, Lemey P, Ping LH, Kouri V, Joanes J, Pérez J, Vandamme AM, Swanstrom R. Limitations to Contact Tracing and Phylogenetic Analysis in Establishing HIV Type 1 Transmission Networks in Cuba . AIDS Res Hum Retroviruses 2007; 23 (3):347–56.
- Pérez L, Pérez Alvarez L. Carmona R. Genotypic Resistence to Antiretroviral drug in patients infected with several HIV type I genetic forms in Cuba. AIDS Res Hum Retroviruses 2007; 23(3):407-14.
- Machado LY, Blanco M, Dubed M, Díaz H, Ruiz N, Valdes N, Romay D, Lobaina L. HIV type 1 genetic diversity in newly diagnosed Cuban patients. AIDS Res Hum Retroviruses 2012; Vol 28 (8): 956-960.
- Aragonés C, Campos JR, Pérez D, Martínez A, Pérez J. SIDATRAT: Informatics to improve HIV/AIDS care. MEDICC Review, 2012; 14 (4): 5–9.
- Nadai Y, Eyzaguirre LM, Constantine NT, Sill AM, Cleghorn F, et al () Protocol for Nearly Full-Length Sequencing of HIV-1 RNA from Plasma. PLoS ONE 2008; 3(1): e1420. doi:10.1371/journal.pone.0001420
- Tamura K, Stecher G, Peterson D, Filipshi A, Kumar S. MEGA 6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 2013; 30:2725-29.
- Guindon S., Dufayard J.F., Lefort V., Anisimova M., Hordijk W., Gascuel O. "New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0." Systematic Biology 2010; 59(3):307-21.
- Anisimova M., Gil M., Dufayard JF., Dessimoz C. and Gascuel O. "Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes." Systematic Biology 2011; 60(5):685-99.
- Anisimova M., Gascuel O. "Approximate Likelihood-Ratio Test for Branches: A Fast, Accurate, and Powerful Alternative." Systematic Biology 2006; 55(4), 539-552.
- Thielen A. Lengauer T. Geno2pheno [454]: A Web Server for the Prediction of HIV-1 Coreceptor Usage from Next-Generation Sequencing Data. Intervirology 2012; 55:113– 117.
- Jensen, M. A., M. Coetzer, A. B. van't Wout, L. Morris, and J. I. Mullins. A reliable phenotype predictor for human immunodeficiency virus type 1 subtype C based on Envelope V3 sequences. J Virol 2006; 80: 4698-4704.
- Machado L, Dubed M, Díaz H, Ruiz N, Romay D, Valdes N, Blanco M, Silva E. Transmitted HIV type 1 Drug Resistence in Newly Diagnosed Cuban Patients. AIDS Res Hum Retroviruses 2013; 29 (2): 411-414.
- Machado LY, Díaz HM, Blanco M, Romay D, Dubed M. Origin and evolutionary history of HIV-1 subtype B in Cuba. MEDICC Review 2017; 19 (2-3): 40-44.
- Machado LY, Díaz HM, Martínez O, Blanco M, Martínez L, Dubed M, Ruiz NM, Silva E. Variantes genéticas del VIH-1, resistencia transmitida a drogas antirretrovirales y progresión clínica en pacientes cubanos no tratados. Revista Panamericana de Infectología 2014; 16 (4): 221-225.
- Blanco M, Machado LY, Díaz HM, Ruiz N, Romay D, Silva E. HIV-1 genetic variability in Cuba and implications for transmission and clinical progression. MEDICC Rev. 2015 Oct; 17(4):25–31.
- Fouchier, R. A., M. Groenink, N. A. Kootstra, M. Tersmette, H. G. Huisman, F. Miedema, and H. Schuitemaker. Phenotype-associated sequence variation in the third variable domain of the human immunodeficiency virus type 1 gp120 molecule. J. Virol 1992; 66: 3183–3187.
- Junqueira DM, de Medeiros RM, Matte MC, Araújo LA, Chies JA, Ashton-Prolla P, et al. Reviewing the history of HIV-1: spread of subtype B in the Americas. PLoS One 2011; 6(11):e27489. DOI: 10.1371/journal.pone. 002 7489.