


|
|
DOI Prefix 10.20431 |
Information
Journal Policies
Change your Toolbox to Create Addiction-Free Opioid Analgesics!
Thomas J. Feuerstein1*, Ulrich G. Hofmann1,2
2.Freiburg Institute for Advanced Studies (FRIAS), University of Freiburg, Germany
Copyright : © 2018 . This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The US opioid crisis stays challenging, as analgesic opioids avoiding both tolerance and addiction are not yet available. The addiction tendency of opioid analgetics may be circumvented by exploiting the principle of biased agonism. Still missing µ-opioid analgetics may be caused by a shortcoming of current agonist’s efficacy estimation. Present efficacy estimates do not yield meaningful correlations with corresponding ligand-biased signaling events. We propose to use the "mechanistic" general response function to deduce the agonist dissociation constant Kd at the receptor under investigation. Accordingly, the transduction shift, pEC50 – pKd, may more reliably estimate the agonist’s efficacy while truly taking spare receptors into account. All in all, the search for adequate opioid replacement drugs should be undertaken with revisited and more suitable tools.
Toolbox, Create Addiction-Free, Opioid Analgesics
1. The Problem
Recently, the current president of the United States has directed the US Department of Health and Human Services to declare the so called “opioid crisis” a public health emergency. Whether or not an overly liberal prescription of opioids in the United States has promoted the epidemic scale of opioid abuse there, is only marginally a pharmacological issue. However, spending money to combat the surge of opioid drug addiction and subsequent casualties in the United States presupposes a better pharmacological understanding of the mechanisms of opioid dependence.
Already in 2013 a panel of leading opioid researchers expressed the hope that opioid agonists with improved tolerance liability could be developed [1]. Thus an intervention path was proposed to combat the most harmful opioid side effects.
However, respective research efforts were undertaken in vain over the last years, as even today opioid agonists avoiding analgesic tolerance or addiction on their own are still not available, because an important functional aspect of opioid receptor regulation is not clearly understood, yet. The contribution of receptor theory to tolerance, addiction and dependence is misjudged. Consequently, the holy grail of present opioid research, i.e. to develop agonists with high analgesic efficacy and low side effects [2] seems unrealistic to achieve with the current approaches.
2. µ-Opioid Receptor-Mediated Responses
Multiple µ-opioid receptor-mediated responses have to be considered. These range from the subcellular environment to the whole organism where the targeted analgesia and the unwanted opioid side effects take place. An overview of most of these µ-opioid receptor responses can be found in [1]. The μ-opioid receptor-activated GTP S binding on the lowest level faces the most complex responses, analgesia, euphoria and/or the disappearance of pain-related dysphoria. On all levels signal transduction mechanisms transform the agonist-induced conformational receptor changes to further signaling events. Seven–transmembrane receptors, like the μ-opioid receptor, can exist in multiple, agonist-dependent active conformations with distinct capabilities to activate effectors [3].
Although the basis by which individual opioids produce different signaling effects is poorly understood [1], examining efficacies of agonists for their biases is mandatory. The concept of efficacy, e.g. the property of a molecule to change a cellular system state, or the ability of an agonist to evoke a receptor response, was further developed to “pluridimensional efficacy” [4]. New molecules may be directed toward clinical testing when the full range of pluridimensional efficacies of to be developed drugs can be utilized. However, with the goal to develop addiction-free drugs one needs a reliable measure of the monodimensional efficacy of each agonist for each response analyzed, i.e. for each dimension of all pluridimensional efficacies.
How can efficacy, the signaling response produced by each drug-receptor-binding event, be quantified? The classical method of Black and Leff [5]fits the agonist concentration-response curve to an “operational” agonist model to assess efficacy and the amount of spare receptors. Efficacies, estimated according to subsequent developments of this method [5], were correlated with receptor-mediated responses when agonists were analyzed for their functional selectivity or ligand-biased signaling [1]. Obviously, it is mandatory that the used efficacy estimates are correct in order to yield meaningful correlations with the receptor-mediated responses under investigation. Unfortunately, however, the present efficacy estimates are faulty (see below).
Biased agonism was defined as the ability of different ligands to differentially activate either signaling cascades or regulatory events. Here, receptor internalization is an important event with respect to addiction as it may diminish spare receptors. Different agonists lead to distinct receptor conformations and potentially internalization. It was suggested to use efficacies as predictors of multiple steps leading to receptor internalization [1]. Opioid agonists not inducing receptor internalization could furnish better chronic analgesics [4]. Receptor internalization of several agonists was linked with their efficacy to identify ligands with low addiction tendency [6]. In Rivero et al. [7], however, only one of sixteen tested agonists displayed the sought bias. We doubt its significance since no α-adjustment of the p value of 0.05 has been made. Clearly, the helpful concept of biased agonism requires improved bias quantifications.
3. Measures Of Agonist Efficacy
Agonist efficacy measures used so far have severe shortcomings and thus the wrong conclusions drawn from these measures may have hindered the needed success in analgesic drug development. Efficacy was described as the signaling response produced by each drug-receptor-binding event [1]. Unfortunately, this definition does not coerce a quantification of the term efficacy. Classically, the operational model [5] or a corresponding method [8] is used as a measure of efficacy (see [4]). Fitting an agonist concentration-response curve according to such an operational model [5] assesses efficacy. Inherently, the operational model aims to reflect the amount of a receptor reserve. However, mentioned methods do not appropriately take into account the discrepancy between concentration-response curves and concentration-binding curves. This discrepancy is intimately linked to spare receptors as represented in the difference between EC50 and Kd of a full agonist.
The following proposes a more suitable way to evaluate agonist efficacies. Correct efficacy estimates should then yield more meaningful correlations with corresponding ligand-biased signaling events in order to learn more about µ-opioid receptor desensitization, tolerance, and addiction risk.
4. Theoretical Background Of Agonist Models
Any function transforming binding into response has to exclude the congruence of a concentration-binding curve and the corresponding concentration-response curve. There is only one exception, the case of direct proportionality between relative binding and relative response ([9], Table 1).

Table1. Direct Proportionality between Relative Binding and Relative Response Take a semi-logarithmic concentration-response curve with Lex being the decadic logarithm lg of the applied concentration (M) of an exogenous agonist which is the independent variable of a function f(Lex). Then, f(Lex), the relative response, is the dependent variable. The semi-logarithmic binding ratio b, describing fractional receptor occupation as function of Lex in the case of a bimolecular reaction, can be deduced from the Law of Mass Action as Kd is the dissociation constant of the agonist at the receptor.
The concentration-response can thus be expressed as a function f(b(Lex)). Then the first derivative of this concentration-response curve, [f(b(Lex))], is not congruent with the first derivative b´(Lex) of a bimolecular binding sigmoid unless there is direct proportionality between receptor occupancy and response:
This inequality only disappears if there is direct proportionality between occupancy and response. Disappearance means that the function f (i.e. the relative response) of a variable (i.e. occupancy) equals that variable and yields f (b (Lex)) = b (Lex). Such a direct proportionality, including parallel shifts in both axis directions, is not possible when spare receptors are present.
Without direct proportionality, e.g. when a receptor reserve exists, the change of the slope function (first derivative) of a concentration-response curve of a full agonist from the slope function of a theoretical binding sigmoid is essential.
The theoretical derivation of a general response function (Table 2) is based on two assumptions [10], adapted here to & micro-opioid receptors:

A functional unit of n μ-opioid receptors may cover only part of the membrane of a presynaptic terminal or an entire postsynaptic cell. Then the activation of all k non-spare receptors of this functional entity results in 100% of the achievable effect from this part of the membrane. When a functional unit covers only part of the membrane, at least two functional units are present.
All functional units in a tissue react similarly to the occupancy of their µ-opioid receptors, e.g. with a reduction in transmitter release presynaptically, or with an appropriate μ-opioid receptor-mediated response postsynaptically.
The occupancy of the n (non-spare and spare) receptors of each functional unit corresponds to a bimolecular reaction.
2. A graded response from 0 to 100% of a functional unit is obtained when none to all of k non-spare receptors are activated. The additional occupation of spare receptors does not contribute.
Then, the activation of exactly k receptors on all functional units would correspond to the maximum effect overall. However, the activation of the fraction k/n of all receptors in the tissue does not result in the activation of exactly k of the n receptors on each functional unit: Some units may have k-1, some others k-2, or k+3, just k, k+1, etc. receptors activated since the activation of the (non-spare and spare) receptors within and between functional units is assumingly independent.
Therefore, the maximum response will not be reached with activation of exactly the fraction k/n of all receptors, since those functional units with less than k receptors activated do not contribute with their maximum effect to the overall effect, and those with more than k receptors activated can only contribute with their maximum effect, but not with more.
Table2. The General Response Function The probability that at least k of n receptors on a functional unit are occupied can be computed from the binomial distribution and equals 1 - probability (0 or 1 or 2 or . . . or k-1 receptors are occupied), which is
Correspondingly, a subset (< 100%) of all functional units contributes with their maximum effect to the overall effect E/Emax. Those functional units with less than k receptors occupied, however, contribute with less than their maximum effect to the overall effect.
Since the probability for such a minor contribution of a unit corresponds to their frequency (or their density) within all the functional units the contribution of these functional units to the overall response E/Emax has to be added as
The factor i/k results in the graded response of these units which is highest for i = k-1, but still lower than the maximum effect of the unit. The combination of the two terms results in the so called general response function:
This function is valid for each binding ratio b (Lex) = 10Lex/ (Kd + 10Lex) between zero and unity.
For k = n the general response function equals the binding ratio, which describes a concentration-response curve in the absence of spare receptors, so response is directly proportional to occupancy. Spare receptors, however, induce a deviation of the shape of a concentration-response curve from the geometrical form of a pure symmetric binding sigmoid for theoretical reasons [11]. Only the binomial distribution appropriately reflects the discrete probability of “success”, i.e. the binding of a ligand at concentration 10Lex M to its receptor in a sequence of independent binding contacts, each asking the binding– no binding question. The probability of each binding event is given by the binding ratio. Therefore, the number i of occupied receptors follows the binomial distribution B [10Lex/(Kd + 10Lex, 1 - 10Lex/(Kd + 10Lex)] at each functional unit. To appropriately model all the binding events, the agonist binding at each single receptor needs to be considered.
The geometrical forms of the general response function for several combinations of n and k are displayed in [10,12,13]. A graphical example is listed below (Fig. 1). Note the asymmetry in the curvature of the curve fitted with the general response function, being broader at low concentrations than at high concentrations. The general response function estimates its parameters from the broader curvature in the range of small concentrations, from the narrower curvature in the range of larger concentrations, and from the steepness of the curve in its asymmetric inflection point. Important characteristics of a receptor reserve are that (1) the agonist concentration of the half-maximal effect, EC50, is lower than its dissociation constant, Kd, and that (2) a submaximal receptor occupancy by a full agonist causes a maximal response [12]. Thus, the general response function sheds light on the meaning of "potency" (represented by EC50), which according to [3], is a “complex function of affinity and efficacy in well coupled tissues”. We think, however, that theEC50is a purely descriptive term, i.e. the location parameter on the abscissa of an agonist-effect curve. The EC50 can easily be estimated by non-linear regression analysis from a cloud of individual concentration-response data points with a logistic function, e.g., in the semi-logarithmic form, E/Emax = 10Lex•c/ (10EC50•c + 10Lex•c) with c being the slope factor (Hill coefficient). The general response function clearly distinguishes affinity (1/Kd) and efficacy, the latter quantifiable as left-shift of the concentration-response curve due to spare receptors in tissues with a relevant amount of a reserve. This left-shift is then lg10Kd– lg10 EC50 = pEC50 – pKd, which we call transduction shift.
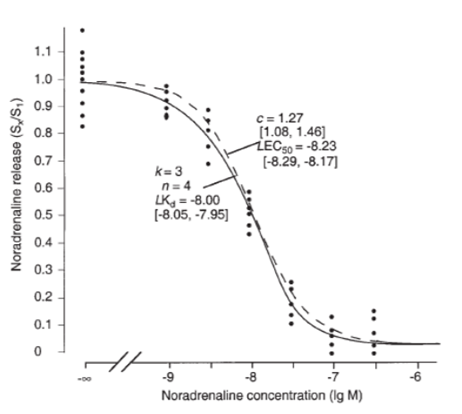
Fig. 7 in [13] reflects an example of such a transduction shift and is displayed here for demonstration (Fig. 1). Nonlinear regression analyses, one with the general response function, the other with a logistic function is shown (Fig. 1). A logistic function is necessarily symmetric in its inflection point. Its abscissa value is the lgEC50. In comparison to the general response function which allows a “mechanistic” interpretation (e.g. a Kd estimate), a logistic function can only have a “descriptive” character (e.g. an EC50 estimate).
The 2-autoreceptor-mediated inhibition by noradrenaline of evoked noradrenaline release in slices of the rabbit hippocampus (for experimental details see [10]).
The dashed inhibition curve was fitted with a logistic function, the solid curve with the general response function. These curves represent the best nonlinear fits to all the individual values (dots) Sx/S 1 (read out of the inhibition of noradrenaline release). The most important parameter estimates together with their CI95s are indicated with the nomenclature -LEC50:= pEC50 and - LKd:= pKd. Estimates of n = 4, k = 3, and of a “true” Kd of 10–8.00 M corresponded to a receptor reserve of 25% [= 100(n – k)/n] which accounted for a significant transduction shift of 0.23 lg units between the EC50 M) and the true Kd of
noradrenaline. We purposely chose an example with a relatively low receptor reserve (25%) to demonstrate the estimation precision of our procedure to evaluate a transduction shift.
The concept of functional units impacts conclusions from functional studies with homogenized material. For instance, a synaptosomal membrane preparation is used to measure a receptor-mediated inhibition of adenylyl cyclase activity. In intact synaptosomes, the receptors under investigation may have a density of n = 10 per functional unit, e.g. a nerve terminal. 60% spare receptors then correspond to k = 4. Membrane homogenization destroys all individual functional units: the homogenate may then be regarded as a single huge functional unit, bearing an extremely high number of receptors, presumably with a preserved receptor reserve of 60%. The activation of 40% of all receptors in the homogenate should then yield 100% of adenylyl cyclase inhibition. However, the activation of 40% of all receptors in intact synaptosomes does not induce a 100% inhibition, since the agonist occupation per synaptosome is not exactly four at each. Only those synaptosomes with four or more occupied receptors contribute with their maximum strength to the overall effect. Therefore, the potency of an agonist will be higher in a homogenate than in intact synaptosomes. Its concentration-response curve is located on the left of that in synaptosomes.
Also in the case of activated ligand-bound complexes the subsequent binding process to an intracellular effector is “binomial”. It reflects the discrete probability of “success”, here the binding of a ligand-bound complex to one of its effectors. If special intracellular effectors exceed the number of currently present ligand-bound complexes these members then represent an effector reserve. Effectors not interacting with ligand-bound complexes may be called spare effectors.
The model of Black and Leff [5] was developed to reflect the above mentioned peculiarities of a receptor reserve, EC50< Kd and maximal response upon submaximal receptor occupancy by a pure agonist. Response expressed by the operational model used a so-called “rectangular hyperbolic” function of the concentration of occupied receptors, called “transducer function”. However, it is fundamentally flawed to suppose a priori that a response function is “rectangular hyperbolic” (for details see [12]). The model ignores the possibility of a relative effect E/Emax to be proportional to the binding ratio. In that case, the agonist dissociation constant Kd is largely misjudged. If there is no direct proportionality between effect and binding ratio, e.g. if a receptor reserve is present, response as function of agonist concentration is not a symmetric sigmoid in semi-logarithmic scale (see above). The operational model does not allow for such a shape and therefore its estimate of a receptor reserve is unbalanced. Additional shortcomings of this model are described in [12].
In summary, the operational model of pharmacological agonism roughly satisfies the pharmacological expectation about a receptor reserve, i.e., EC50< Kd. This is achieved, however, without relying on the physico-chemical grounds of pharmacology since the “transducer ratio” or the “operational efficacy” of an agonist (see [5]) has no clear biological meaning. The price to be paid for the realization of EC50< Kd is that the operational model may detect a receptor reserve which does not exist (see [9, 12]).
Spare receptors must induce a change of the first derivative of a concentration-response curve of a full agonist from that of a binding sigmoid [9]. This condition has not been considered by Black and Leff [5]. Consequently, the operational model does not yield appropriate efficacy estimations and may have hindered so far the discovery of ligand-biased signaling events pointing to addiction-free opioid drugs.
How should one proceed if a decision has to be made between a descriptive [5] and a mechanistic model[10]? Nonlinear regression analysis provides an objective criterion, the goodness-of-fit, i.e. the Residual Sums of Squares (RSS). We recommend the following pragmatic approach to decide between competing models: To prefer a mechanistic model over a descriptive model, the mechanistic model should yield at least an equivalent quality of the fit procedure if both models have nearly the same number of parameters to be estimated. The number of parameters of the logistic function (Emax, EC50, c) appears less than the number of parameters of the general response function (Emax, Kd, n, k).The Hill coefficient c is a real variable and n and k are positive integer variables. The fraction of spare receptors is estimated by the quotient (n–k)/n. The quotient thus may represent a single “real” parameter, corresponding to the single “real” c. With the same number of parameters of the two models, “equivalence” may be assumed if the percentage difference of the RSS values is less than 10%.
Most agonists don´t simply mimic endogenous ligands but cause receptors to exercise only portions of their vast repertoire of behaviors [3]. This receptor-based selectivity or biased agonism represents an expansion of the activation possibilities of an endogenous agonist. The corresponding targeting of chemical structures of ligands as possibility to induce a selective cellular function may be available avenue for therapeutic selectivity.
Obviously, such a therapeutic progress is urgently needed in the development of new µ-opioid receptor analgesic drugs which should lose their addiction risks. Obtaining accurate measurements of agonist efficacy is most important in the analysis of opioid tolerance (e.g.[6]). Ligand-related conformation changes as biased agonism at the µ-opioid receptor were correlated with agonist efficacies to activate receptor internalization [6]. However, unexpectedly, some µ-opioid receptor agonists did not display a good correlation between the operational efficacy for G protein activation and the operational efficacy for the activation of receptor internalization. Are these discrepancies due to the use of the operational model of Black and Leff [5] to estimate efficacies? May other implementations to discriminate between agonist affinity and efficacy yield better results?
We think that the above-mentioned transduction shift, pEC50 – pKd according to the general response function, is a better tool than the operational efficacy.
To (partly) explain the ability of opioids to induce tolerance three theories were proposed .[6]: (1) Low efficacy µ-opioid agonists induce profound tolerance because they give rise to receptor desensitization, but little receptor internalization. Then the receptor is unable to undergo efficient dephosphorylation and resensitization. (2) Morphine differs from other µ-opioid agonists by different molecular mechanisms to desensitize µ-opioid receptors.
(3) The low efficacy of morphine induces little µ-opioid receptor desensitization. Its prolonged signaling at the receptor leads to neuronal adaptations that precipitate tolerance.
All three theories may be compromised by agonist efficacy estimates with shortcomings. Refinements of the operational model .[5] cannot solve that and probably have hindered to find biased µ-opioid replacement drugs with low or no addiction tendency. It seems advisable to explore other ways of analyzing agonists.
We recommend to evaluate the inflection point asymmetry and the steepness at the inflection point of concentration-response curves by nonlinear regression analysis, keeping the following in mind: (1) the data point resolution on the lg concentration abscissa should be as high as possible. It is more important to sample the concentration space with many data points than scarce data points with many measurements. The minimal concentration difference must not exceed a half lg-unit. Dense sampling of the concentration scale improves nonlinear fits. (2) Test concentrations should include extremal (low and high) values to cover the flat branches of the curve. The zero concentration of the tested ligand should lie at least one lg-unit below the lowest real ligand concentration. Without sampling these flat tails, the deviations from symmetry of fitted curves cannot be recognized. (3) Nonlinear regression analyses must be based on the individual data points, not on their means.
Thus, it is advisable to sample the concentration space in the widest possible way and to use fit models suitable for also detecting asymmetry in concentration-response curves. For this purpose it may be necessary to supplement concentration-response curves used for previous analyzes.
Transduction shifts, pEC50 – pKd according to the general response function, should be the applied efficacy estimates, replacing the method of Black and Leff [5]. All in all, the long path to adequate replacement drugs to effectively combat the opioid crisis has only just began, but should be undertaken with a more suitable set of tools, including new considerations (Table 3). The use of incorrect estimation methods for the efficacy of agonists must be avoided.
Table3. Binding duration as a key to biased agonism?
With respect to the principle of biased agonism, the use of different chemical structures of ligands as alternatives to induce selective cellular functions, both the spatial dimensional accuracy of an agonist regarding the µ-opioid receptor binding pocket and its actual conformation deserves attention. We propose to analyze the principle of biased agonism of µ-opioid receptor ligands also with respect to their binding duration at the receptor systems under investigation.
The prolonged signaling of a morphine-activated receptor points to the possible relevance of the ligand-receptor interaction duration. The binding duration can be evaluated rather easily and has been shown to be crucial to the distinction of full and partial agonists .[14]: The loss of efficacy of partial agonists is most likely related to a shorter binding duration as compared to full agonists. In a spare- receptor-free system it became obvious that partial agonists do not bind long enough to the receptor to mediate a maximum response[14]. Since the binding duration of a ligand cannot depend on the density of receptors, i.e. on the possible existence of a receptor reserve, the determination of the binding duration of µ-opioid receptor ligands may help to elucidate their role in biased agonism. Correspondingly, such spare receptor-free conditions may be established by the use of irrevesrsible µ-opioid receptor antagonists (see .[7]). Then, partial and full agonists can be easily distinguished their different binding durations may linked to differently selected cellular functions.
Acknowledgement
We thank Nancy Campbell of FRIAS to funnel our pharmacological interest to the ongoing opioid crisis.
References
- Williams JT, Ingram SL, Henderson G, Chavkin C, von Zastrow M, Schulz S, Koch T, Evans CJ, Christie MJ (2013) Regulation of μ- opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev 65:223-254
- Morgan MM, Christie MJ (2011) Analysis of opioid efficacy, tolerance, addiction and dependence from cell culture to human. Br J Pharmacol 164:1322-1334
- Kenakin T (2007) Functional Selectivity through Protean and Biased Agonism: Who Steers the Ship? Mol Pharmacol 72:1393-1401
- Kenakin T (2011) Functional Selectivity and Biased Receptor Signaling. J Pharmacol Exp Ther 336:296-302
- Black JW, Leff P (1983) Operational models of pharmacological agonism. Proc R Soc B 220:141–162
- McPherson J, Rivero G, Baptist M, Llorente J, Al-Sabah S, Krasel C, Dewey WL, Bailey CP, Rosethorne EM, Charlton SJ, Henderson G, Kelly E (2010) μ-Opioid receptors: correlation of agonist efficacy for signalling with ability to activate internalization. Mol Pharmacol 78:756-766
- Rivero G, Llorente J, McPherson J, Cooke A, Mundell SJ, McArdle CA, Rosethorne EM, Charlton SJ, Krasel C, Bailey CP, Henderson G, Kelly E (2012) Endomorphin-2: a biased agonist at the μ-opioid receptor. Mol Pharmacol 82:178-188
- Ehlert FJ (1985) The relationship between muscarinic receptor occupancy and adenylate cyclase inhibition in the rabbit myocardium. Mol Pharmacology 28:410-421
- Agneter E, Singer EA, Sauermann W, Feuerstein TJ (1997) The slope parameter of concentration-response curves used as a touchstone for the existence of spare receptors. Naunyn-Schmiedeberg's Arch Pharmacol 356:283-292
- Feuerstein TJ, Sauermann W, Allgaier C, Agneter E, Singer EA (1994) New insights into receptor theory, as provided by an artificial partial agonist made-to-measure. Naunyn- Schmiedeberg´s Arch Pharmacol 350:1-9
- Feuerstein TJ, Sauermann W, Allgaier C, Singer EA (1993) Mathematical modelling and quantification of the autoinhibitory feedback control of noradrenaline release in brain slices. Naunyn-Schmiedeberg's Arch Pharmacol 347:171-179
- Sauermann W, Feuerstein TJ (1998) some mathematical models for concentration- response relationships. Biometrical J 40, 7:865- 881
- Feuerstein TJ, Limberger N (1999) Mathematical analysis of the control of neurotransmitter release by presynaptic receptors as a supplement to experimental data. Naunyn-Schmiedeberg´s Arch Pharmacol 359: 349-359
- Hoeren M, Brawek B, Mantovani M, Löffler M, Steffens M, van Velthoven V, Feuerstein TJ (2008) Partial agonism at the human 2A-autoreceptor: Role of binding duration. Naunyn-Schmiedeberg´s Arch Pharmacol 378:17-26